Nanoporous palladium with sub-10 nm dendrites by electrodeposition for ethanol and ethylene glycol oxidation†
Serhiy
Cherevko
*,
Nadiia
Kulyk
and
Chan-Hwa
Chung
*
Advanced Materials and Process Research Center for IT, School of Chemical Engineering, Sungkyunkwan University, Suwon, 440-746, Republic of Korea. E-mail: s.cherevko@gmail.com; chchung@skku.edu; Fax: +82 31-290-7272; Tel: +82 31-290-7260
First published on 9th November 2011
Abstract
High surface area Pd foams with roughness factors of more than 1000 and a specific surface area of 60 m2 g−1 are obtained by electrodeposition. The foams are composed of dendrites with branches on the 10 nm scale. The resulting electrodes show high activity towards the oxidation of C2alcohols.
“Palladium is a remarkable metal”—wrote Jiro Tsuji, the author of several books and reviews about the use of palladium in organic synthesis, in his recent book.1 It is one of the most important and widely used catalysts in organic synthesis and pharmaceutics.1a,2 In fuel cell technology, palladium is considered as a promising material for hydrogen storage and for hydrogen purification from a feed stream. It is also useful in the area of fuel cell catalysts and hydrogen detection and sensing.3 Together with hydrogen, low molecular weight alcohols can also be used as a fuel in such cells. One of the major problems in current fuel cell technology is the sluggish kinetics of oxidation of the fuels. In this regard, researchers have started to look back to the alkaline direct alcohol fuel cell (ADAFC) technology, which has been renewed in recent years. Palladium, which shows the highest catalytic activity for the ethanol oxidation reaction (EOR) in alkaline media among pure metals, is considered as one of the most promising candidates as a catalyst in ADAFCs.4
For many of the applications described above, highly dispersed, high-surface-area (HSA) palladium is preferable. Several methods have been proposed for the preparation of HSA nanoporous Pd, such as dealloying,5electrodeposition using organic and inorganic templates,6 and combustion synthesis.7 However, most of these methods are time consuming and, with several exceptions, result in the preparation of palladium with a large amount of impurities, e.g. the presence of less noble metals in the Pd sponges formed by dealloying.5
Recently, the utilization of the hydrogen evolution reaction (HER) in the formation of HSA self-supported metals was shown.8 We reported the preparation of Ag and Au foams.9 However, the application of the same technique to Pd resulted in the formation of Pd films composed of relatively bulky particles.10 Herein, we show that, at very high current densities, the formation of highly dispersed bimodal micro- and nano-porous palladium foams can be achieved.
Nanoporous palladium can be deposited from a simple solution containing 3.75–30 mM PdCl2 and 1 M H2SO4 at a potential E = −4 V vs.Ag/AgCl and deposition time of 30 s.
For the applied deposition conditions, films deposited from a solution with 15 mM of Pd2+ show the most appropriate morphology (multilayered foam without cracks). These foams are very uniform over the entire electrode area (microscope field-of-view of ca. 2.4 × 1.8 mm2) (Fig. S1a and b, ESI†). A slightly magnified view is presented in Fig. 1a. On the nanoscale, the foam is composed of tiny dendrites (Fig. S1c, ESI†). The TEM image presented in Fig. 1b reveals the presence of a highly dispersed, relatively uniform network of Pd dendrites. The width of the dendrite branches is less than 10 nm, as shown in the HR-TEM image in Fig. 1c. The interplanar spacing measured for the stem and two side branches matches well with the value of d{111} = 0.2458 nm in the reference Pd sample (JCPDS 046-1043). The orientations of the crystal planes suggest that the growth direction is along the [111] axis. The XRD and EDS data (Fig. S2a and b, ESI†) reveal the chemical purity and crystal structure of the Pd foam.
 | ||
| Fig. 1 (a) SEM, (b) TEM, and (c) HR-TEM images of the nanoporous palladium obtained from the solution containing 15 mM of Pd2+. | ||
As we showed before in the case of the deposition of nanoporous Ag and Au,9a,11 a large amount of metal ions is crucial for the formation of such honeycomb-like structures. When the amount of Pd2+ in the solution grows from 3.75 mM to 15 mM, the deposition rate increases and relatively thick honeycomb-like structures form on the electrode, though with some cracks when the amount of Pd ions is too high (Fig. S3a–d, ESI†). When the applied overpotential is very high, the deposition of Pd is controlled by the diffusion of Pd ions within the diffusion layer. Both the concentration and diffusion layer thickness, δ, affect the mass transfer coefficient. The decrease of the latter is obtained by the convection created by the detachment of the hydrogen bubbles and their movement to the electrolyte–air interface. The very high current density of the HER (ca. j = 3–5 A cm−2) leads to δ values of a few micrometres.12 It should be noted that during the deposition of Pd from the electrolyte based on NH4Cl, the current density was only j = 0.75 A cm−2 (E = −4 V).10 This may explain the difference in the morphologies. The amount of Pd deposited is proportional to the concentration of Pd ions in the solution (Table S1, ESI†).
The results shown herein appear to contradict our previous statement that only normal and intermediate metals can form relatively small dendrites during hydrogen evolution assisted electrodeposition.9a The current data, however, suggest that at even higher rates of hydrogen evolution, inert metals can form dendrites. The decrease in δ leads to the increase in the diffusion limited current, which decreases the initiation overpotential. Consequently, our current results reveal the possibility of preparing highly dispersed foams of other inert metals (Fe, Ni, Co, etc.).
One of the most important parameters of HSA materials is the roughness factor (Rf). The active electrochemical surface area, SESA, may be measured by analysing the palladium voltammetric profile in an acidic media (Fig. 2a) Here, three potential regions corresponding to oxygen adsorption/desorption, double layer (DL) charging, and hydrogen sorption/desorption are highlighted. Such a CV profile is characteristic of highly dispersed palladium and differs significantly from that of conventional bulk Pd. On a bulk palladium electrode, electrochemical H absorption (Habs) into Pd, H adsorption (Hads), and HER take place at the same potentials and resolving these processes is virtually impossible.13 The voltammogram from such an electrode is usually similar to that observed on porous Pd composed of relatively big particles (Fig. 4a in ref. 10). Hence, the SESA, for palladium, is usually obtained by analysing the oxygen region. The charge obtained from DL charging is also used to estimate the SESA of metals,14 and particularly that of Pd.15 In contrast to the massive palladium, the Habs overpotential is larger for dispersed systems.16 Also, the hydrogen diffusion length is significantly shorter than that of thick film electrodes. Thus, the adsorption and desorption of hydrogen from the surface of palladium (peaks C1,C2 and A2,A1, respectively) can be clearly separated from the bulk processes (C3/A3). A similar voltammetric profile in the Hads region was also observed on mesoporous Pd.6a,dFig. 2b presents the variation of Rf with the amount of Pd2+ in the electrolyte. The data for Rf were obtained by three methods: using charges of hydrogen and oxygen adsorption and capacitance in the double layer. The three methods show the same gradual increase in surface area with increasing amount of Pd2+ in the solution. It should be noted that the capacitance of the double layer used herein is not 20 μF cm−2, as is usually assumed.14,15 The value of 45 μF cm−2 was chosen as it provides the best fit to the data obtained by the two other methods. Assuming that the surface area estimated from the oxygen region is correct, the H coverage can be obtained from the relationship between the Rf values found from the oxygen and hydrogen regions. The calculated H coverage was ca. 0.85 ± 0.02 (at E = −0.1 V vs.Ag/AgCl) for the four electrodes, which is higher than the value of 0.76 found for a thin (a few tens of nanometres) layer of Pd on Au(111), though the authors used a different reference value for the hydrogen adsorption charge and the coverage was measured at slightly different potential.15 It is not clear, however, if this is because the hydrogen does not fully cover palladium surface or whether some hydrogen was desorbed from the surface simultaneously with its desorption from the bulk. The specific surface area was estimated to be ca. 60 m2 g−1 (Table S1, ESI†), which is lower than that of H1-e palladium films6d but much higher than that of nanoporous Pd prepared by dealloying (23 m2 g−1),17Pd nanowires synthesized in hexagonal mesophases (12 m2 g−1),18 and the values for palladium black reported in the literature (17–23 m2 g−1).18
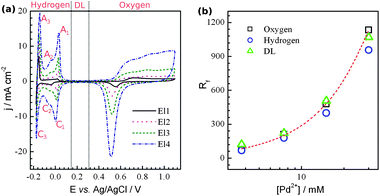 | ||
| Fig. 2 (a) Cyclic voltammograms in 1 M H2SO4 taken on films deposited from the solution containing the following amounts of Pd2+ in mM: 3.75 (El1), 7.5 (El2), 15 (El3), and 30 (El4) at 5 mV s−1. (b) Dependence of the nanoporous Pd roughness factor on the concentration of Pd2+ in the electrolyte used for deposition. | ||
To evaluate the performance of the nanoporous Pd electrode as a cathode for ADAFCs, the electrooxidation of two high energy density fuels, i.e.ethanol (EtOH) and ethylene glycol (EG), was performed. The results are shown in Fig. 3. (The full scans for EG are shown in Fig. S4, ESI†). It can be seen that EtOH and EG oxidation currents increase with increasing Rf of the electrode. The results obtained are, however, very different from those reported in the literature.19 In our case, for all of the electrodes, the presence of at least two peaks in the alcohols' oxidation region on forward going (anodic) scans can be seen. For El1, the first peak (peak I) is relatively low, but becomes larger and more pronounced with increasing surface area. Usually, higher concentrations are used in the study of the oxidation of alcohols. Fig. 3c and d present the CVs taken on the same electrodes but in solutions with alcohols having concentrations of 0.1–0.3 M. The addition of alcohols to the solution results in an increase of both peak currents. Beside the increase in current, the peak potentials are shifted to more positive values. As a result, the voltammetric profile of 0.3 M EtOH oxidation shows only a small shoulder at potentials corresponding to peak II. If the EtOH concentration were further increased, peak I would shift further, resulting in voltammograms similar to those reported in the literature (Fig. S5, ESI†). It is known that the oxidations of both EtOH and EG are very complex processes which may proceed via several consecutive and/or parallel steps involving different reaction intermediates.19b The results obtained suggest that peaks I and II result from the oxidation of the adsorbed and solution species, respectively. It is most likely that the change in the Rf of the electrode changes the relative currents of different steps in the oxidation of the alcohols. In the case of EtOH oxidation, the change in the peak I and peak II currents suggests the involvement of the surface for the former and diffusion control (also Pd oxidation) for the latter. Thus, as Rf increases, only a slight enhancement of the peak II current is observed. On the other hand, peak I increases gradually with increasing Rf. In the case of EG oxidation, however, both peaks increase gradually with increasing Rf. It is clear that additional experiments on the mechanism of the oxidation process are needed.
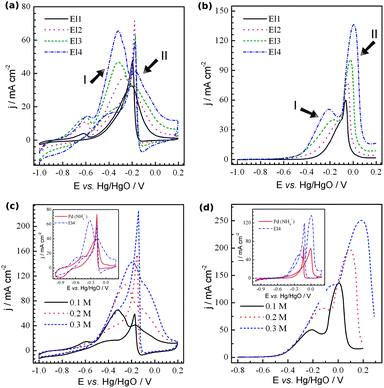 | ||
| Fig. 3 Cyclic voltammograms recorded with nanoporous Pd electrodes in (a) 1 M NaOH + 0.1 M ethanol and (b) 1 M NaOH + 0.1 M ethylene glycol. (c) and (d) show the CVs taken with El4 in 1 M NaOH with the amount of (c) ethanol and (d) ethylene glycol indicated. The insets present the comparison of the oxidation of the corresponding alcohols on electrodes El3 and porous Pd deposited from the NH4Cl based electrolyte. The scan rate was 20 mV s−1. | ||
We also performed additional experiments on the oxidation of the alcohols, but on porous Pd with relatively big particles (El5) (whose morphology is similar to that shown in Fig. 1b and b′ in ref. 10). This electrode is more accessible for the electrolyte (the ratio between the alcohol accessible area and real surface area is higher). The comparison is shown in the insets of Fig. 3c and d. The voltammetric profiles of both EtOH and EG oxidation on El5 are similar to those in the literature. The EtOH and EG oxidation peaks correspond to peak II on electrodes 1–4, and those on El5 do not show any significant current increase at potentials corresponding to peak I. The pseudo-steady-state curves of EtOH oxidation taken at varying potentials show the similar difference in the current–voltage dependence profiles (Fig. S6a and b, ESI†). EG oxidation shows much higher currents on El3 (even when they are normalized by the real surface area (Fig. S6d, ESI†)) at low potentials (E < −0.2 V vs. Hg/HgO), suggesting that there is an increase in the material activity. This activity may be related to the higher probability of the intermediates trapped within such a highly porous structure being oxidized. In terms of its gravimetric activity, nanoporous Pd shows extremely high currents even at the relatively low alcohol concentration of 0.1 M that is utilized, especially for the films with a low Rf where the effect of the mass transfer limitation of the active species to internal layers of the electrode is minimized, i.e.EtOH and EG oxidation peak currents of 221 and 413 A g−1 on El1 (Table S1, ESI†).
In conclusion, Pd foams were prepared by hydrogen evolution assisted electrodeposition. Due to their high surface area and non-uniform accessibility of the reagents, such foams show unusual voltammetric profiles. We believe that the extremely high surface area of ca. 60 m2 g−1 and oxidation activity of the C2alcohols (peak currents as high as Ip,EtOH = 221 A g−1 and Ip,EG = 413 A g−1) make porous Pd a promising material in such important areas as heterogeneous catalysis, fuel cells, and sensors.
This research was supported by the Basic Science Research Program (2011-0010546) and Gyeonggi Province through the GRRC program in Sungkyunkwan University.
Notes and references
- (a) J. Tsuji, Palladium in Organic Synthesis, Springer, Berlin, 2005 Search PubMed; (b) R. Mahrwald, Adv. Synth. Catal., 2007, 349, 472 CrossRef.
- J. Tsuji, Palladium Reagents and Catalysts: New Perspectives for the 21st Century, John Wiley & Sons, Ltd, Chichester, 2004 Search PubMed.
- S. Cherevko, N. Kulyk, J. Fu and C.-H. Chung, Sens. Actuators, B, 2009, 136, 388–391 CrossRef.
- (a) E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431–3450 CrossRef CAS; (b) C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS.
- J. Yu, Y. Ding, C. Xu, A. Inoue, T. Sakurai and M. Chen, Chem. Mater., 2008, 20, 4548–4550 CrossRef CAS.
- (a) G. Denuault, C. Milhano and D. Pletcher, Phys. Chem. Chem. Phys., 2005, 7, 3545–3551 RSC; (b) S. Cherevko, J. Fu, N. Kulyk, S. M. Cho, S. Haam and C.-H. Chung, J. Nanosci. Nanotechnol., 2009, 9, 3154–3159 CrossRef CAS; (c) P. Bartlett and J. Marwan, Phys. Chem. Chem. Phys., 2004, 6, 2895–2898 RSC; (d) P. N. Bartlett, B. Gollas, S. Guerin and J. Marwan, Phys. Chem. Chem. Phys., 2002, 4, 3835–3842 RSC.
- B. C. Tappan, S. A. Steiner and E. P. Luther, Angew. Chem., Int. Ed. Engl., 2010, 49, 4544–4565 CrossRef CAS.
- S. Wen and J. A. Szpunar, Micro Nano Lett., 2006, 1, 89–93 CAS.
- (a) S. Cherevko and C.-H. Chung, Electrochem. Commun., 2011, 13, 16–19 CrossRef CAS; (b) S. Cherevko, X. Xing and C.-H. Chung, Electrochem. Commun., 2010, 12, 467–470 CrossRef CAS.
- X. Xing, S. Cherevko and C.-H. Chung, Mater. Chem. Phys., 2011, 126, 36–40 CrossRef CAS.
- S. Cherevko and C.-H. Chung, Electrochim. Acta, 2010, 55, 6383–6390 CrossRef CAS.
- N. Nikolic, K. Popov, L. J. Pavlovic and M. Pavlovic, J. Electroanal. Chem., 2006, 588, 88–98 CrossRef CAS.
- M. Grden, M. Lukaszewski, G. Jerkiewicz and A. Czerwinski, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, INC., Hoboken, 2nd edn, 2001 Search PubMed.
- H. Duncan and A. Lasia, Electrochim. Acta, 2008, 53, 6845–6850 CrossRef CAS.
- H. Duncan and A. Lasia, Electrochim. Acta, 2007, 52, 6195–6205 CrossRef CAS.
- X. Wang, W. Wang, Z. Qi, C. Zhao, H. Ji and Z. Zhang, Electrochem. Commun., 2009, 11, 1896–1899 CrossRef CAS.
- F. Ksar, G. Surendran, L. Ramos, B. Keita, L. Nadjo, E. Prouzet, P. Beaunier, A. Hagège, F. Audonnet and H. Remita, Chem. Mater., 2009, 21, 1612–1617 CrossRef CAS.
- (a) C. W. Xu, H. Wang, P. K. Shen and S. P. Jiang, Adv. Mater., 2007, 19, 4256–4259 CrossRef CAS; (b) V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Marchionni, F. Vizza, L. Q. Wang and P. K. Shen, Fuel Cells, 2010, 10, 582–590 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental conditions, additional SEM images, CVs, pseudo-steady-state curves, and summary table. See DOI: 10.1039/c1nr11316j |
| This journal is © The Royal Society of Chemistry 2012 |