Controlled synthesis of radiolabelled amine methacrylate water-soluble polymers with end-groups of varying hydrophobicity and studies of adsorption behaviour†
Mark
Long
a,
David W.
Thornthwaite
a,
Suzanne H.
Rogers
a,
Francis R.
Livens
b and
Steve P.
Rannard
*c
aUnilever Research and Development Port Sunlight Laboratories, Quarry Road East, Bebington, Wirral, UK CH63 3JW
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, UK M13 9PL
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: srannard@liv.ac.uk; Fax: +44 151 794 3501; Tel: +44 151 794 3501
First published on 4th November 2011
Abstract
The effect of chain end chemistry on the behaviour of poly(2-(diethylamino)ethyl methacrylate) has been studied through the placement of various radioactive groups with increasing hydrophobicity at the chain end of the polymer. Controlled radical polymerisation with 14C-labelled initiators was used to form polymers of very similar chain length including doubly-labelled, fluorescent-radio polymers. Monitoring and quantification of behaviour was conducted purely using radiotechniques, without the need for fluorescence measurement, showing a clear impact on solid surface adsorption across a range of surface types (filter paper, photographic paper and hair). The studies presented here have clear implications for the study of polymer behaviour through low level fluorescent modification of water-soluble polymers.
Introduction
Radioisotopes have been used for many years to study chemical, biological and physical processes and elucidate reaction mechanisms.1–8 Predominantly for reasons of cost and safety, alternative methods to label and monitor materials and molecules have been developed with the use of fluorophores and fluorescence spectroscopy being especially common.9,10 In the context of polymer science, fluorescence studies have added mechanistic and physical insight to mixed polymer-surfactant systems,11 diffusion in gels and solids,12polymerisation mechanism,13–19 and polymer behaviour in solution.20–29When fluorophores are chemically bound to a polymer to aid detection and monitor behaviour, there is an implicit assumption that the fluorophore does not significantly affect the properties of the polymer and that the observations effectively report the behaviour of the unlabelled materials. As the molecular weight and relative dimensions of the labels can be significant and the fluorophores are often hydrophobic, the validity of this assumption in the case of water-soluble polymers requires careful investigation. The introduction of aromatic fluorophores can be considered as a hydrophobic modification leading to an amphiphilic polymer11 with subsequent modification of behaviour such as solubility, association and surface interactions. In a 1996 review of his own work and published data from other groups regarding the behaviour of fluorescently labelled poly(methacrylic acid) in aqueous environments, Morawetz19 concluded that the published behavioural models based on fluorescent labelling were valid for the labelled polymer only and “cannot be used to infer the state of the unlabeled” poly(methacrylic acid). Best and Silescu29 concluded, from fluorescence densitometry at polymer-polymer interfaces, that the effects of fluorophore addition in polymer blend studies could be reduced by ensuring that the labelled polymers contained at least 200 monomer units per fluorescent label. Egan et al.16 found that increasing the number of fluorophores on poly((2-ethyl hexyl) methacrylate) chains used as stabilizers in the dispersion polymerization of poly(vinyl acetate) resulted in changes to the mean particle size, distribution, composition and molecular weights of the final colloidal polymer particles when compared to stabilisers without fluorescent modification. Increased fluorophore addition within the stabilizer resulted in reduced mean particle sizes and large amounts of irreversibly attached poly((2-ethyl hexyl) methacrylate). The particle size distribution was also affected with the addition of an average of just one fluorophore per stabilizer molecule leading to the appearance of an ultra-high molecular weight polymer fraction.
It is clear from the literature examples presented that the inclusion of a fluorophore can have substantial effects on the physical and chemical properties of polymers and assumptions of identical or near-identical behaviour of modified and unmodified materials are not necessarily valid. It is also well-established that the addition of even small numbers of hydrophobic groups, fluorescent or non-fluorescent, to water-soluble polymers can affect behaviour at interfaces,30a in blends and in solution and this has been utilised in many industrial applications such as associative thickeners.30b
The use of radiolabels in combination with fluorescent labels uniquely provides an independent measure of effects arising from the presence of a fluorophore without relying on fluorescence measurement. As already mentioned, radioisotopes have been widely used to monitor chemical processes and early reports of their use in polymer science date to the 1950s. The Bevington and Ayrey groups pioneered radioisotope application predominantly in the investigation of free radical polymerization mechanism;31–35 however, aspects of polymer stability, including bio-stability of polyurethanes,36poly(methyl methacrylate)hydrolysis,37 and stabiliser mobility within polymers38 have also been investigated. In medicine, radiolabelled materials have been used to investigate the in vivo behaviour of polymers used to deliver drugs and assist in the treatment of cancer.39–44 The environmental biodegradability of synthetic aliphatic polymers such as poly(acrylic acid), poly(methacrylic acid), poly(sodium acrylate), poly(ethyl acrylate-co-methacrylic acid), poly(3-hydroxybutyrate-co-3-hydroxyoctanoate) and poly(ε-caprolactone) have also been studied.45–49 Physical properties such as the kinetics of adsorption at liquid–liquid interfaces51 and the adsorption/desorption and distribution of polyelectrolytes and uncharged polymers at solid-liquid interfaces such as glass,50silica,52 leather,53poly(tetrafluoroethylene), PTFE,54 and surface modified polystyrene latex particles55 have previously been explored using radiolabelling techniques.
Although polymer deposition/adsorption has been determined using either radio55–57 or fluorescent55,58,59 labels, very few studies have directly compared radiolabelled material with identical fluorescently labelled materials55 or controllably combined both labels within one polymer chain, thereby employing doubly-labelled materials.
Using a 14C radioisotope to investigate the physicochemical properties of a polymer does not change the hydrophilicity/hydrophobicity, charge or molecular structure and has a negligible impact on molecular weight. Providing such labelled polymers are radiochemically stable on the timescale of the experiment, combining fluorescent labels with 14C radiolabels in a systematic manner allows the investigation of the effect of hydrophobic and fluorophore modification on the polymer chemical and physical behaviour without the inherent issues of fluorescer quenching.60
Our recent reports have highlighted the synthesis of 14C-labelled 2-bromoisobutyric acid61a,b and its use in monitoring radiolabelled initiators during and after Atom Transfer Radical Polymerisation (ATRP). We have also drawn attention to the importance of small changes in chain end chemistry (even a single methyl group) in controlling the solution behaviour of water-soluble amine methacrylates, specifically the lower critical solution temperature of poly(2-(dimethylamino)ethyl methacrylate).61c
Herein we report a strategy to controllably introduce radioactivity at the chain end of a water-soluble polymer generated through ambient temperature ATRP using an 2-propanol/watersolvent mixture and Cu(I)Br/2,2′-bipyridinecatalyst system.62 We utilise 14C radiolabelled initiators with increasing hydrophobicity and ultimately generate a combined radio/fluorescent-labelled polymer containing one radioactive site and a single fluorophore (9-hydroxyfluorene)63 at the chain end, Fig. 1. The adsorption of poly(2-(diethylamino)ethyl methacrylate) (pDEAEMA) from aqueous solution onto various substrates is monitored and the effect of the varying chain-end hydrophobicity is studied.
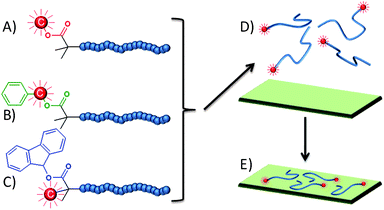 | ||
| Fig. 1 Radiolabelled polymers with different end groups; A) methyl, B) benzyl and C) 9-hydroxyfluorene terminated. D) Labelled polymers in aqueous solution and E) adsorption onto surfaces. | ||
Results and discussion
1. Radio and fluorescent-labelled initiator and polymer design, synthesis and characterisation
Three initiators were synthesised using two approaches; either conventional ester synthesis utilising the reaction of 2-bromoisobutyryl bromide, 1, with 14C-labelled primary alcohols, 2 or 3, or the reaction of 14C-labelled 2-bromoisobutyric acid, 4, with 1,1-carbonyl diimidazole, 5, to generate an acid imidazolide which is capable of esterification with hydroxyl functional materials. In this second approach 9-hydroxyfluorene, 6, was utilised thereby generating a doubly-labelled radio-fluorescent initiator, 9. The syntheses are summarised in Scheme 1.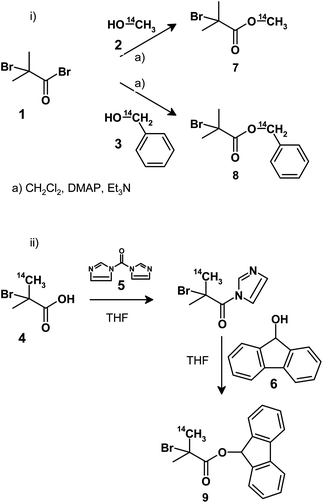 | ||
| Scheme 1 Synthesis of 14C-radiolabelled ATRPinitiators: i) reaction with 14C-labelled primary alcohols and ii) reaction of 14C-labelled 2-bromoisobutyric acid with 9-hydroxyfluorene. | ||
The synthesis of radio-labelled materials requires assessment of both chemical and radio purity as labelled impurities will considerably hamper the accurate identification and quantification of the target material. The initiators were therefore subjected to radio thin layer chromatography (R-TLC)60a,b under different solvent conditions. Each initiator was accurately assessed using different ratios of 40–60 petroleum ether and diethyl ether; 7 utilised a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 v/v ratio whilst 8 and 9 were analysed using a 90:10 v/v solvent mixture, Fig. 2. Analysis of the initiators using 1H and 13C nuclear magnetic resonance spectroscopy (NMR) (see Electronic Supporting Information, ESI) and UV-vis spectrophotometry was comparable to the analysis of unlabelled analogue materials. Collectively the analysis demonstrated a high chemical and radiochemical purity for the initiators.
:40 v/v ratio whilst 8 and 9 were analysed using a 90:10 v/v solvent mixture, Fig. 2. Analysis of the initiators using 1H and 13C nuclear magnetic resonance spectroscopy (NMR) (see Electronic Supporting Information, ESI) and UV-vis spectrophotometry was comparable to the analysis of unlabelled analogue materials. Collectively the analysis demonstrated a high chemical and radiochemical purity for the initiators.
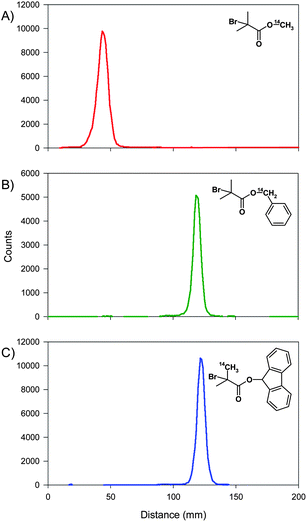 | ||
| Fig. 2 Radio thin layer chromatography of 14C-labelled initiators: A) methyl 2-bromoisobutyrate, 7 (eluent: 60:40 petroleum ether 40–60/Et2O v/v), B) benzyl 2-bromoisobutyrate, 8 (eluent: 90:10 petroleum ether 40–60/Et2O v/v), and C) 9-hydroxyfluorenyl 2-bromoisobutyrate, 9 (eluent: 90:10 petroleum ether 40–60/Et2O v/v). | ||
The non-radiolabelled analogue initiators were used to demonstrate the ability of each material to polymerise the amine containing monomer 2-(N,N-diethylamino) ethyl methacrylate (DEAEMA) under identical ambient ATRP conditions using a 90/10 v/v 2-propanol/watersolvent mixture. The formation of p(DEAEMA) was compared with the identical polymerisations utilising the 14C-labelled initiators, Table 1. The presence of the radiolabel had no appreciable effect on the outcome of the polymerisation or the number average degree of polymerisation (DPn) as measured by 1H NMR or gel permeation chromatography (GPC). Although there are differences in the observed Mn and DPn, as measured by either GPC or 1H NMR, the GPC results were determined using only refractive index and a standard calibration curve generated from poly(methyl methacrylate) standards, therefore the GPC data is reported as poly(methyl methacrylate) equivalent molecular weights. The three radiopolymers were considered sufficiently comparable within each measurement to utilise in studies of end-group effects.
An evaluation of the kinetics of the polymerisations showed slightly different rates of reaction when using each initiator and greater than 96% conversion of DEAEMA in each case, Fig. 3. Although the polymerisations are not ideal, the methyl-functionalinitiator, 7, led to a slower polymerisation than the benzyl-functionalinitiator8, which in turn was slower than the 9-hydroxyfluorenyl-functionalinitiator9.
![Comparative kinetic evaluation of the ambient polymerisation of 2-(N,N-diethylamino)ethyl methacrylate with initiators7 (red data), 8 (green data) and 9 (blue data). Conversion is shown as coloured circles and ln[M0]/[M] is given as triangles. Guidelines are shown to emphasise the differences of initiation and propagation.](/image/article/2012/PY/c1py00397f/c1py00397f-f3.gif) | ||
| Fig. 3 Comparative kinetic evaluation of the ambient polymerisation of 2-(N,N-diethylamino)ethyl methacrylate with initiators7 (red data), 8 (green data) and 9 (blue data). Conversion is shown as coloured circles and ln[M0]/[M] is given as triangles. Guidelines are shown to emphasise the differences of initiation and propagation. | ||
Residual unreacted initiator was quantified by R-TLC through the elution of the final polymer sample with a 90/10 v/v mixture of petroleum ether and acetic acid (see ESI) as previously described.60a,b Radioactivity associated with unreacted initiator varied from 3.72% (initiator7) to 7.02% (initiator8) and 14.59% (initiator9) suggesting a decreasing initiator efficiency but also following the trend of increasing polymerisation rate i.e. faster polymerisation resulting in higher levels of unreacted initiator. Although this seems counterintuitive (fewer radicals should maintain a slower polymerisation rate), these findings agree with our previously reported examination of initiator consumption under ATRP conditions,60b and the identified continual formation of new chains at monomer conversions >90% and polymerisation times >200 min. The rapid consumption of monomer would result in a higher unreacted initiator concentration within the final polymer sample if relatively slow initiation (i.e. initial addition of monomer to unreacted initiator) relative to propagation is present during the polymerisation, possibly related to initiator structure or solubility. For truly ‘living’ polymerisation to be observed, the rate of initiation must be considerably higher than the rate of propagation64 (and termination). ATRP is a ‘controlled radical’ process and not a ‘living’ polymerisation (mistakenly described as such in many reports). Within the ATRPpolymerisations that we have studied with radiolabelled initiators, truly ‘living’ conditions have not been observed as expected; however, linear semi-logarithm plots suggest the attainment of a steady propagating radical concentrations during the main stages of propagation, even with considerable unreacted initiator present. Further work will focus on additional kinetic studies, however this is not the focus of this report.
2. Quantification of polymer adsorption onto hair, cellulose filter paper and photographic paper
As discussed previously, fluorescent modification is a common approach to study the solution, solid state and adsorption behaviour of polymers. Fluorescent labels allow the rapid identification and quantification of modified polymers to elucidate temporal, gravimetric and spatial information. Difficulties arise when the distribution of fluorophore is not homogeneous across the sample, i.e. some polymer chains have high fluorophore modification, or the fluorophore interferes with the analysis technique, e.g. self-quenching.The polymers of the current study have been designed to vary only in chain-end modification, from methyl to benzyl to 9-hydroxyfluorenylgroups, and specifically to carry a single radioactive carbon at each chain-end to allow quantification of the behaviour of each polymer irrespective of fluorescence. Three substrates were chosen to establish variability of adsorption under identical controlled conditions using commercially relevant and non-ideal surface chemistry; untreated ‘virgin’ hair, filter paper and photographic paper were used as models of biomaterials (keratin), hydrophilic cellulosic and hydrophobically modified substrates respectively (see ESI).
To model a conventional experimental procedure, the radiolabelled polymers were purified by flash column chromatography to remove catalyst and ligand residues, dried to remove all traces of reaction solvent, and subsequently used to prepare a 0.14% w/w solution at pH 2. The radioactivity of the solution was determined by liquid scintillation counting to provide initial activity levels. The papers were cut into 2 cm2 samples whilst standard 5 cm virgin hair swatch samples were used as received.
Two radioanalytical techniques, storage phosphor imaging, Fig. 4, and liquid scintillation counting were employed, with multiple repeats, to study each sample.
 | ||
| Fig. 4 Autoradiography (storage phosphor imaging) images of radiolabelled poly(2-(diethylamino)ethyl methacrylate) adsorbed onto A) hair, B) filter paper and C) photographic paper. Poly(2-(diethylamino)ethyl methacrylate) has varying initiator-derived end-groups: i) methyl, ii) benzyl and iii) 9-hydroxyfluorenyl. Experiment-relevant autoradiography standards are shown for comparison. | ||
Storage phosphor imaging is an autoradiography technique allowing visualisation and quantification of radioactivity compared against radioactive standards. Sample oxidation, and subsequent liquid scintillation counting, is a destructive technique that measures the total radioactivity of the substrate after exposure. Five sample substrates were suspended in solutions containing radiolabelled polymer for up to 48 h with periodic removal for analysis. All samples were allowed to drain before air drying for 24 h prior to analysis.
Storage phosphor imaging of the hair samples, Fig. 4Ai–iii, allowed only a qualitative examination of adsorption as the samples were asymmetric and varied in thickness. Shielding of β-emission from the 14C nucleus (path length = 0.28 mm in water/tissue) also prevents accurate measurement using this technique. Even with these limitations for the hair samples, it was visually clear that higher radioactivity was present when polymers containing the 9-hydroxyfluorenyl end group, Fig. 4Aiii, were present in solution, than those comprising the benzyl end group, Fig. 4Aii, or the methyl end group, Fig. 4Ai.
A similar trend could be visually determined within the adsorption of the radiolabelled p(DEAEMA) onto filter paper, Fig. 4Bi–iii, and photographic paper, Fig. 4Ci–iii; however, it was most noticeable in the adsorption onto photo paper. Although accurate determination of polymer adsorption onto hair could not be accomplished by autoradiography, the filter paper and photographic paper samples were quantified to determine the mass of polymer adsorbed/surface area over the 48 h of the experiment, Fig. 5.
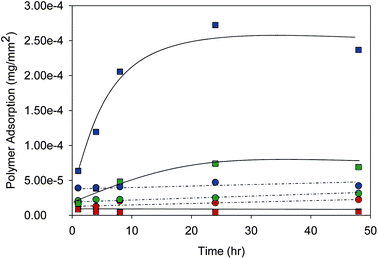 | ||
| Fig. 5 Adsorption of radiolabelled poly(2-(diethylamino)ethyl methacrylate) onto filter paper (circles) and photographic paper (squares) measured by autoradiography. Initiator-derived end-group variation includes methyl (red), benzyl (green) and 9-hydroxyfluorenyl (blue) groups. Adsorption was conducted at pH 2 and ambient temperature. | ||
P(DEAEMA) adsorption was shown to be both substrate specific and end-group directed. Although the differences of polymer adsorption onto filter paper were relatively small, the same trend of adsorption (with respect to end-group) was observed across filter paper and photo paper; 9-hydroxyfluorenyl > benzyl > methyl. Adsorption to photographic paper was higher than filter paper for benzyl and 9-hydroxyfluorenyl end-groups. The presence of the methyl end-group did not appear to direct preferential adsorption onto the photographic paper; however, the porosity of the filter paper may account for the observed differences.
A second measure of adsorption, not affected by porosity, utilised complete sample oxidation with collection/capture of 14CO2 (3-methoxypropylamine) and liquid scintillation counting. Samples of the three substrates were subjected to this destructive evaluation technique and the adsorption of polymer was determined as a mass of polymer/mass of substrate. Comparison of the adsorption of polymers containing different end-groups is shown in Fig. 6.
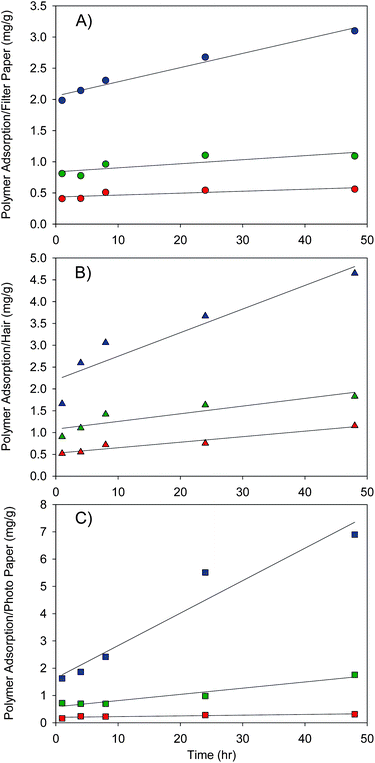 | ||
| Fig. 6 Adsorption of radiolabelled poly(2-(diethylamino)ethyl methacrylate) onto A) filter paper (circles), B) ‘virgin’ hair (triangles) and C) photographic paper (squares) as measured by oxidation and liquid scintillation counting. Initiator-derived end-group variation includes methyl (red), benzyl (green) and 9-hydroxyfluorenyl (blue) groups. Adsorption was conducted at pH 2 and ambient temperature. | ||
As observed with autoradiography, within each substrate-specific data set, the order of adsorption was identical with respect to polymer chain-end: 9-hydroxyfluorenyl > benzyl > methyl group. The presence of the most hydrophobic chain-end (9-hydroxyfluorenylgroup) considerably enhanced the surface adsorption on all substrate types, generally increasing the adsorbed mass by a factor of >3 over the methyl end-group. It is questionable therefore whether the modification of water-soluble polymers with hydrophobic fluorescent dyes to study adsorption is valid experimentally, especially when additional complications of fluorescer quenching are also considered.
Although the data clearly show the impact of a single modification to the polymer chain-end, and the effect of increasing the hydrophobicity of the modification, Fig. 6 compares chain-ends within a single substrate. Additional comparisons were made for each chain-end (ESI) to determine whether the specific chains-ends were directed preferentially towards each substrate. Within the methyl-modified and benzyl-modified polymer adsorption data it is clear that although different behaviour is seen for each substrate, the overall adsorption of p(DEAEMA) is not particularly high, with a maximum value of approximately 1.8 bmg g−1 for the benzyl-modified polymer adsorbing onto hair and photographic paper after 48 h. The methyl- and benzyl-modified polymers both follow a clear trend of adsorption for exposure times; hair > filter paper > photographic paper. There is no clear difference in absolute material adsorption after 48 h for the benzyl modification onto hair and photographic paper, and substrate specificity of the methyl or benzyl chain-end modification is therefore only suggested by these data; however, it is well demonstrated within the 9-hydroxyfluorenyl modified polymers.
The introduction of the 9-hydroxyfluorenyl end group leads to adsorption to photographic paper being enhanced over adsorption to both hair (∼1.5×) and filter paper (∼2x). Also, the magnitude of adsorption to each substrate of the 9-hydroxyfluorenyl-terminated polymer is considerably higher than the other end-groups studied; ∼3× increase in adsorption to filter paper over benzyl end-group modification (∼5.5× over methyl end-group); ∼2.5× increase in adsorption to hair over benzyl chain ends (∼4× over methyl modification); ∼4× increase in adsorption to photographic paper over the benzyl modification (>20× increase over methyl modification). The values of adsorbed polymer presented are much higher than would be expected by single-layer deposition and are more comparable to those achieved through layer-by-layer desposition,65 indicating uncontrolled multiple-layer deposition or complex behaviour of the non-ideal uncharacterised substrates at pH 2. Recent studies of p(DEAEMA), and other water-soluble polymers, with hydrophobic end groups in acidic water, have shown unexpected self-assembly and aggregation.66 Such self-assembly may also account for enhanced deposition/adsorption within the polymers studied here.
3. Surface tensiometry
Polymer chain-end modification also has the potential to alter the air–water surface interactions of the polymers, therefore the non-radioactive polymers were studied using surface tensiometry. p(DEAEMA) is a relatively hydrophobic polymer and was not highly soluble in water. 2% w/w solutions of the three non-radioactive polymers were therefore prepared in pH2buffer solution and diluted twelve times, each time to half of the previous concentration. Eight repeated surface tension measurements were made for each dilution and the mean surface tension was plotted against polymer concentration, Fig. 7. The maximum concentrations achievable for each polymer were not high enough to determine the critical micelle concentration (CMC); however, the surface tension decreased with increasing polymer concentration, as would be expected with an amphiphilic polymer in solution. The polymers derived from 7 and 8 behaved similarly; however, polymers derived from the 9-hydroxyfluorenylinitiator, 9, generated a steeper decrease in surface tension with increasing polymer concentration. As the CMC values were not determined, the C10 values (concentration of polymer required to decrease the water surface tension by 10 mN m−1) were obtained through examination of the negative gradient of the curves (see ESI); C10 (w/w) = 0.62% (methyl), 0.56% (benzyl) and 0.45% (9-hydroxyfluorenyl). The chain-end directed increase in polymer hydrophobicity was clearly evident. C10 values were chosen as these represent the midpoint of the decrease in surface tension observed from a 2% (w/w) solution of each polymer (i.e. a maximum of approximate 20 mN m−1 decrease was observed across the solutions). The same trend was seen for C5, C15 and C20 values; however, the C20 values have been extrapolated from the available data (C20 (w/w), extrapolated = 3.09% (methyl), 2.66% (benzyl) and 1.93% (9-hydroxyfluorenyl)).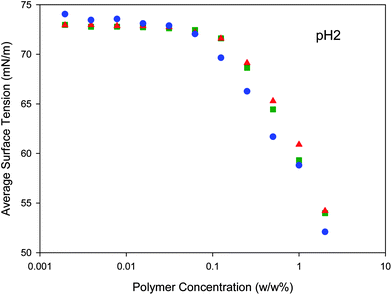 | ||
| Fig. 7 Surface tensiometry of poly(2-(diethylamino)ethyl methacrylate) with different chain-end modification; methyl (red triangles), benzyl (green squares) and 9-hydroxy fluorenyl (blue circles) at pH2. | ||
Conclusions
The use of analogous radiolabelled and non-radiolabelled atom transfer radical polymerisationinitiators has allowed the synthesis of a range of readily detectable poly(2-(diethylamino)ethyl methacrylate) polymers with similar chain lengths. We have shown that small differences in the initiator-derived chain end chemistry can significantly affect adsorption of the amine-methacrylatepolymer from aqueous solutions onto various substrates, apparently driven by the relative hydrophobicity of the polymer chain end. Although this finding is somewhat to be expected, the relative impact of each chain-end has been shown to vary greatly between surface types. Extrapolation of polymer behaviour to new substrates therefore cannot be readily achieved and the impact of hydrophobic modification cannot be ignored when studying polymer behaviour with new substrates. All modifications of water-soluble polymers would benefit from initial evaluation using chemistry-independent methods, e.g. radioactivity rather than fluorescent techniques to ascertain the role of the fluorescer in directing polymer behaviour. Assembly at the air/water interface was also observed to vary depending on chain-end chemistry.We chose the most hydrophobic chain-end within our study as a fluorescent label, used by many researchers to establish the solution and surface adsorption behaviour of water-soluble polymers. Clearly, our radioactivity studies quantify the effect of controlled placement of just one fluorescent group per polymer chain for p(DEAEMA), thereby questioning the assumptions that small numbers of large hydrophobic fluorescent groups have little effect during polymer behaviour measurement. This is even more important in fluorescent labelling experiments where the label is incorporated statistically (i.e. through reaction onto pendant functional groups or the polymerisation of fluorescent monomers) as it is probable that many chains contain more than one fluorescent label.
Acknowledgements
The authors wish to thank the Royal Society for an Industry Fellowship (SR), Unilever for financial support (ML) and Dr Ben Carter of the Centre for Materials Discovery for aid during surface tension studies. Rob Rannard is also warmly thanked for help compiling graphical data.Notes and references
- G. Lappin and S. Temple, Radiotracers in Drug Development, CRC Press, 2006 Search PubMed.
- D. Dalvie, Curr. Pharm. Des., 2000, 6, 1009 CrossRef CAS.
- J. E. Casida, Residue Rev., 1969, 25, 149 Search PubMed.
- C. J. Collins, Adv. Phys. Org. Chem., 1964, 2, 3.
- J. C. Bevington, Adv. Polym. Sci., 1960, 2, 1 CrossRef CAS.
- W. W. Cleland, J. Labelled Compd. Radiopharm., 2007, 50, 1006 CrossRef CAS.
- E. A. Evans, Synthesis and Applications of Isotopically Labelled Compounds 1991, Proceedings of the International Symposium, Ed. E. Buncel and G. W. Kabalka, Elsevier Science, 1992 Search PubMed.
- T. J. Ruth, Rep. Prog. Phys., 2009, 72, 1.
- J. R. Wenner and V. A. Bloomfield, Anal. Biochem., 1999, 268, 201 CrossRef CAS.
- K. Haupt, A. G. Mayes and K. Mosbach, Anal. Chem., 1998, 70, 3936 CrossRef CAS.
- F. M. Winnik and S. T. A. Regismond, Surf. Sci. Series, 1998, 77, 267 Search PubMed.
- L. Masaro and X. X. Zhu, Prog. Polym. Sci., 1999, 24, 731 CrossRef CAS.
- S. Wroblewski, K. Trzebiatowska, B. Jedrzejewska, M. Pietrzak, R. Gawinecki and J. Paczkowski, J. Chem. Soc., Perkin Trans. 2, 1999, 1909 RSC.
- M. Mertoglu, A. Laschewsky, K. Skrabania and C. Wieland, Macromolecules, 2005, 38, 3601 CrossRef CAS.
- M. Spiniello, A. Blencowe and G. G. Qiao, J. Polym. Sci., 2008, 46, 2422 Search PubMed.
- L. S. Egan, M. A. Winnik and M. D. Croucher, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 1895 CrossRef CAS.
- S. Sosnowski, J. Feng and M. A. Winnik, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1497 CrossRef CAS.
- C. S. Kim, S. M. Oh, S. Kim and C. G. Cho, Macromol. Rapid Commun., 1998, 19, 191 CrossRef CAS.
- H. Morawetz, Macromolecules, 1996, 29, 2689 CrossRef CAS.
- I. Souter and L. Swanson, Macromolecules, 1994, 27, 4304 CrossRef CAS.
- G. A. Baker, A. N. Watkins, S. Pandey and F. V. Bright, Analyst, 1999, 124, 373 RSC.
- J. Kevelam, S. Martinucci and J. B. F. N. Engberts, Langmuir, 1999, 15, 4989 CrossRef CAS.
- T. Sasaki, H. Arisawa and M. Yamamoto, Polym. J., 1991, 23, 103 CrossRef CAS.
- H. Morawetz, Science, 1988, 240, 172 CrossRef CAS.
- W. B. Wang, J. H. Ali, R. B. Dorshow, M. A. McLoughlin and R. R. Alfano, SPIE International Society Optical Engineering, 1999, 3600, 227 Search PubMed.
- F. W. Wang and R. E. Lowry, Polymer, 1985, 26, 1046 CrossRef CAS.
- H. Morawetz, Makromol. Chem., 1985, 13, 67 CrossRef CAS.
- H. J. Egelhaaf, E. Holder, P. Herman, H. A. Mayer, D. Oelkrug and E. Lindner, J. Mater. Chem., 2001, 11, 2445 RSC.
- M. Best and H. Sillescu, Polymer, 1992, 33, 5245 CrossRef CAS.
- (a) J. Hedin, A. Oestlund and M. Nyden, Carbohydr. Polym., 2010, 79, 606 CrossRef CAS; (b) G. Fonnum, J. Bakke and F. K. Hansen, Colloid Polym. Sci., 1993, 271, 380 CAS.
- G. Ayrey, Chem. Rev., 1963, 63, 645 CrossRef.
- G. Ayrey, D. Barnard and T. H. Houseman, Chem. Rev., 1971, 71, 372.
- J. C. Bevington and J. R. Ebdon, Developments in Polymerization 2nd Edition, Ed. R. N. Haward, Applied Science, London, 1979 Search PubMed.
- J. C. Bevington, Trends Polym. Sci., 1993, 1, 68 CAS.
- C. A. Barson, J. C. Bevington and B. J. Hunt, J. Macromol. Sci., Part A: Pure Appl. Chem., 2000, A37, 1261 Search PubMed.
- R. S. Labow, D. J. Erfle and J. P. Santerre, Biomaterials, 1995, 16, 51 CrossRef CAS.
- J. C. Bevington, R. Brinson and B. J. Hunt, Makromol. Chem., 1970, 134, 320.
- T. J. Henman and S. R. Oldland, Dev. Polym. Stabilization, 1984, 7, 275 Search PubMed.
- M. Kissel, P. Peschke, V. Subr, K. Ulbrich, J. Schuhmacher, J. Debus and E. Friedrich, J. Pharm. Sci. Tech., 2001, 55, 191 Search PubMed.
- D. Domurado, P. Fournie, C. Braud and M. Vert, J. Bioact. Compat. Polym., 2003, 18, 23 CrossRef CAS.
- F. A. Dorkoosh, M. P. M. Stokkel, D. Blok, G. Borchard, M. R. Tehrani, J. C. Verhoef and H. E. Junginger, J. Controlled Release, 2004, 99, 199 CrossRef CAS.
- E. R. Gillies, E. Dy, J. M. J. Fréchet and F. C. Szoka, Mol. Pharmaceutics, 2005, 2, 129 CrossRef CAS.
- N. Nasongkia, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842 CrossRef CAS.
- M. Hruby, J. Kucka, H. Mackova, C. Konak, M. Vetrik, J. Kozempel and O. Lebeda, J. Appl. Polym. Sci., 2009, 111, 2220 CrossRef CAS.
- T. W. Federle, M. A. Barlaz, C. A. Pettigrew, K. M. Kerr, J. J. Kemper, B. A. Nuck and L. A. Schechtman, Biomacromolecules, 2002, 3, 813 CrossRef CAS.
- S. Ponsart, J. Coudane, B. Saulnier, J. L. Morgat and M. Vert, Biomacromolecules, 2001, 2, 373 CrossRef CAS.
- K. M. Jop, P. D. Guiney, K. P. Christensen and E. M. Silberhorn, Ecotoxicol. Environ. Saf., 1997, 37, 229 CrossRef CAS.
- M. B. Freeman and T. M. Bender, Environ. Technol., 1993, 14, 101 CrossRef CAS.
- C. E. Warburton, J. R. Flynn and L. T. Flynn, J. Appl. Polym. Sci., 1972, 16, 1235 CrossRef CAS.
- A. Voronov, S. Minko, S. Alexander and E. Pefferkorn, Colloid Polym. Sci., 2004, 282, 1000 CrossRef CAS.
- R. Wang and J. B. Schlenoff, Macromolecules, 1998, 31, 494 CrossRef CAS.
- O. Oulanti, J. Widmaier, E. Pefferkorn, S. Champ and H. Auweter, J. Colloid Interface Sci., 2005, 291, 98 CrossRef CAS.
- K. Pietrucha and J. Kroh, Radiat. Phys. and Chem., 1985, 25, 451 Search PubMed.
- L. Ouali and E. Pefferkorn, Macromolecules, 1996, 29, 686 CrossRef CAS.
- S. Ponsart, J. Coudane, J. L. Morgat and M. Vert, J. Labelled Compd. Radiopharm., 2001, 44, 677 CrossRef CAS.
- A. R. Sykes and P. A. Hammes, Drug Cosmet. Ind., 1980, 62 Search PubMed.
- J. Woodard, J. Soc. Cosmet. Chem., 1972, 23, 593 Search PubMed.
- H. D. Weigmann, Y. K. Kamath, S. B. Ruetsch, P. Busch and H. Tesmann, J. Soc. Cosmet. Chem., 1990, 41, 379 Search PubMed.
- S. T. A. Regismond, Y. M. Heng, E. D. Goddard and F. M. Winnik, Langmuir, 1999, 15, 3007 CrossRef CAS.
- B. Gole, S. Shanmugaraju, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 10046 RSC.
- (a) M. Long, D. W. Thornthwaite, S. H. Rogers, G. Bonzi, F. R. Livens and S. P. Rannard, Chem. Commun., 2009, 6406 RSC; (b) M. Long, S. H. Rogers, D. W. Thornthwaite, F. R. Livens and S. P. Rannard, Polym. Chem., 2011, 2, 581 RSC; (c) S. Jana, S. P. Rannard and A. I. Cooper, Chem. Commun., 2007, 2962 RSC.
- (a) S. McDonald and S. P. Rannard, Macromolecules, 2001, 34, 8600 CrossRef CAS; (b) T. He, D. J. Adams, M. F. Butler, C. T. Yeoh, A. I. Cooper and S. P. Rannard, Angew. Chem., Int. Ed., 2007, 46, 9243 CrossRef CAS; (c) T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495 CrossRef CAS.
- I. Toulokhonova, B. Bjerke-Kroll and R. West, J. Organomet. Chem., 2003, 686, 101 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168 CrossRef CAS.
- S. A. Sukhishvili and S. Granick, Macromolecules, 2002, 35, 301 CrossRef CAS.
- J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental details, radio-characterisation, 1H NMR spectra of initiators, GPC characterisation of p(DEAEMA), R-TLC of polymers, regression curves derived from surface tension measurements. See DOI: 10.1039/c1py00397f |
| This journal is © The Royal Society of Chemistry 2012 |