Injectable biodegradable polymeric system for preserving the active form and delayed-release of camptothecin anticancer drugs†
Olcay
Mert ‡
a,
Güneş
Esendağlı
b,
A. Lale
Doğan
b and
Ayhan S.
Demir
*a
aDepartment of Chemistry, Middle East Technical University, 06531, Ankara, Turkey. E-mail: asdemir@metu.edu.tr; Tel: 0090 (0)312 210 32 42
bDepartment of Basic Oncology, Institute of Oncology, Hacettepe University, 06100, Ankara, Turkey. Tel: 0090 (0)312 305 44 06
First published on 27th October 2011
Abstract
One of the most challenging problems for camptothecin (CPT) family anticancer drugs (i.e. topotecan (TPT)) is the conversion of the active lactone ring into an inactive toxic carboxylate form under physiological conditions (pH = 7.4) in the body. Therefore, a simple platform based on thermosensitive PLLA-mPEG gels was designed to maintain TPT and CPT in lactone form, especially for brain tumor therapies. A high stabilization of the lactone species CPT and TPT within gel (>95%), efficient versatile homogenous drug loadings at 0.015%, 1%, and 10%, and the sustained-release of CPT and TPT over three weeks were all successful. The stabilization mechanism of drugs with gel was elucidated by ATR-FTIR, confocal and light microscopy. The cytotoxic efficacy of TPT in the PLLA-mPEG platform (PLLA-mPEG-TPT) was evaluated on LLC-1 and 4T1 cancer cell lines. In vivo, the administration of PLLA-mPEG-TPT to mice with breast tumors resulted in a significant reduction in tumor size and better survival percentages.
Introduction
The development of a method for the administration of drugs has great importance, especially for the effective treatment of patients with cancer. It represents a major challenge. The technologies that are designed to “control release” bioactive agents fall into relatively small categories. New sustained drug delivery systems are being explored to overcome the disadvantages of conventional dosage forms. Numerous strategies to reach this goal have been explored. These strategies frequently have been based on the use of ‘biodegradable polymeric biomaterials’.1–5 The release rate of a drug must be controlled in order to maintain a therapeutically desirable concentration, avoid unfavorable side effects, and be compatible with the surrounding environment. A controlled release formulation would overcome the drawbacks of traditional drug therapy, such as short exposure time, rapid clearance, and the need for multiple injections.6–8 It may also reduce toxicity due to the reduced dose.Injectable and biodegradable block copolymer drug delivery systems have advantages over sustained drug delivery systems. Some of these polymers have the unusual property of gelling upon heating. The resulting materials are hydrogels and they are injectable because in an aqueous environment they exhibit temperature dependent reversible gel-sol transitions. As shown by Jeong et al., PLLA-PEG block copolymer is a liquid at around 45 °C and it is injectable in that form.1 When it is injected, it forms into a gel and releases the bioactive drug into its surroundings.1 These controlled release biocompatible hydrogel systems have advantages for local drug delivery; e.g., around the solid tumor site. These biodegradable injectable controlled release systems can also be easily used for the treatment of brain tumors. The reasoning here is that the blood barrier limits drugs and/or excess drugs.
Camptothecins anticancer drugs possess α-hydroxy-δ-lactone ring functionality. They are a new class of drugs that target the enzyme topoisomerase.9 Camptothecins selectively target topoisomerase I while etoposide inhibits topoisomerase II.9 This property is lost under physiological conditions (pH 7 or above). The lactone moiety readily opens and yields the inactive carboxylate form, and is converted into the form of ionic species (Fig. 1).10–15 This ionic species is formed rapidly when it is exposed to a basic environment, including blood, pH 7 or above. This ionic form is not only inactive but also very toxic to animals and humans.
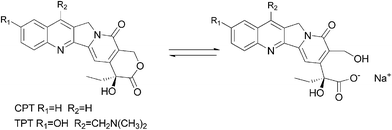 | ||
| Fig. 1 Camptothecins hydrolysis to water soluble sodium salt. | ||
In the work by Gao et al.,16 9-nitro-20(S)-camptothecin (9-NC) was loaded into micelles formed from copolymer methoxy poly(ethylene glycol)-b-poly(D,L-lactide) (mPEG-PDLLA, PELA). The lactone form of 9-NC in the micellar solution decreased up to 80% at 160 min, and the curve probably goes down beyond 160 min. On the other hand, 9-nitro-20(S)-camptothecin (rubitecan or Orathecin) had its New Drug Application (NDA) withdrawn by the FDA due to the treatment of pancreatic cancer patients who failed at least one prior chemotherapy regimen. Finally, there was no evidence as to how the stabilized drugs in micelles worked in a cell culture or an in vivo model application. Yu et al.17 recently published a study centered on the use of a PEG-CPT drug, which was released from hydrogels formed by a thermally gelling and hydrolytically degradable polymer PLGA-PEG-PLGA. The disadvantage is the poor performance of commercial pergylated CPT in clinical Phase 2b trials in patients with gastric or gastroesophageal cancers whose disease progressed following prior chemotherapy. Last, at the in vivo application of the report, the performance of the PEG-CPT was evaluated towards 5-fluorouracil (5-Fu) as the positive control group. Thus, it is unclear whether chemically modified pegylated-CPT gives a better response than CPT itself or not. Berrada et al.18 prepared a chitosan based autogelling solution with camptothecin, which remains a liquid at low temperature and turns into gel when heated. It was claimed that approximately 80% of the intact drug was loaded in chitosan gels, although lactone–carboxylate conversion was not experimentally reported in the initial solution and latter gel state. In addition, a gel-CPT preparation was tested by administering it directly onto the tumor mass and differently compared its efficacy with the CPT solution given intraperitoneally.
In the current study, the activity loss of camptothecin family anticancer drugs was prevented with a simple efficient platform using injectable and biodegradable block copolymer PLLA-mPEG gels. In vivo, the application of the platform confirmed that the polymeric system is much more effective than the free drug itself.
Materials and methods
L-Lactide (Aldrich), mPEG-2000 homopolymer (Aldrich), and stannous 2-ethyl-hexanoate (Sigma) were used in the syntheses of PLLA-mPEG diblock copolymers. Diethyl ether (J.T. Baker) and dichloromethane (J.T. Baker) were employed in the purification steps. Trisma-base (Sigma), sodium hydroxide (Fisher), and hydrochloric acid (Fisher) were used in the buffer preparations. Camptothecin and topotecan were purchased from Chengdu Haojie Pharmchem in China and used as received. Triethylamine (J.T. Baker), acetonitrile (J.T. Baker), and acetic acid (J.T. Baker) were employed in the preparation of the mobile phase and filtered out with a filtration system prior to use.1H and 13C NMR experiments were carried out with Bruker Avance DPX 400 for the structural analysis and determination of the number average molecular weight of the copolymers. The attenuated total reflection FTIR spectroscopy (ATR-FTIR) was a Bruker Vertex 70, equipped with the PIKE MIRacle universal ATR sampling accessory (Diamond/ZnSe crystal plate with a fixed angle of incidence of 45 with 2.0 μ depth penetration). HPLC spectra were recorded on Thermo Finnigan HPLC with a Fluorescence detector. Data was analyzed with the Chromquest software program. Nova-Pak C-18 4 μm 3.9 × 150 mm HPLC column was used for the analysis of carboxylate and lactone forms of anticancer drugs. Fluorescence measurements were recorded on a Perkin Elmer LS-50 fluorescence spectroscopy. Double wavelength and single read programs with Win-Lab software were used for the analysis of lactone and carboxylate conversions and the release experiments of anticancer drugs. Measurements were performed in a quartz cell. Zeiss LSM 510 Confocal Microscope equipped with an Argon laser (488 nm) and LP505 emission filter was employed. 40× magnification was performed. Zeiss Axioskop 2 plus optic microscope with 100× magnification with a Hitachi CCD camera was used for the CPT loaded and unloaded gel.
Copolymerization of L-lactide with mPEG-2000 (PLLA-mPEG2k)
PLLA-mPEG2k diblock copolymers were synthesized with a similar methodology that we described previously.19 Briefly, for the synthesis of 1, 1.2 g of mPEG-2000 (0.6 mmol), 5 g of L-lactide (35 mmol), and 100 mg of Sn(Oct)2 (0.25 mmol) were added to 120 mL of dry toluene and refluxed for 10.5 h under nitrogen atmosphere with a Dean–Stark apparatus. The flask was brought to room temperature. Then, toluene was evaporated under reduced pressure. The copolymer product was dissolved in 100 mL dichloromethane and precipitated with an excess amount of cold diethyl ether (1 L). 1H NMR (CDCl3) δ: 1.5 (3H, d), 3.63 (2H, s), 5.1 (H, q); 13C NMR (CDCl3) δ 16.6, 68.9, 71, 169.3.PLLA-mPEG2k 2 and 3 were synthesized with the same methodology as above. Experimental conditions and molecular weight data were shown in Tables 1 and 2. 1H-NMR spectra for copolymers 1, 2, and 3 were indicated in the section of “Copy of 1H-NMR spectra” in the ESI.†
Determination of the gel to sol transition temperature
Diblock PLLA-mPEG copolymers 1–3 were mixed with distilled water to make various concentrations. They were vortexed in order to obtain a homogeneous mixture, if they were capable, and they were immersed in a temperature controlled water bath. The gel-to-sol transitions of copolymers were examined from 20 to 80 °C in increments of 2.0 °C. The tubes were maintained in water baths for 5 min at each temperature point before tilting. The critical gel-to-sol temperature was determined as the temperature that caused the gel to turn into the sol form immediately after the tilting of the tube.1CPT and TPT loading into copolymer gel
0.015, 1.0, and 10% loadings of topotecan and camptothecin into copolymer gel 3 were performed (Table 3). For example, the 0.015% loading of CPT into copolymer was prepared in the following way: 100 mg PLGA-mPEG, 0.015 mg CPT, and 200 μL of tris buffer at pH: 7.4 were mixed to form gel at RT. For the measurement of <1 mg CPT and TPT; 1–2 mg of drugs were dissolved in the mixture of MeOH: DCM. Then, the appropriate volume of drug solution, corresponding to 0.015 mg in weight, was placed into a 1 mL Eppendorph tube followed by evaporation with a rotavap and lyophilizator overnight for the full removal of solvents. For higher loadings, camptothecin or topotecan powders were directly mixed with the copolymer. Then, the appropriate volume of buffer was added to form gels.Preparation of tris buffer
606 mg Trisma-base was added to 250 mL of distilled water. Then, it was stirred for complete dissolution. The medium of pH was adjusted to 5.0, 7.4, or 9.9 with diluted hydrochloric acid or sodium hydroxide solutions before filtration with a filter unit.Preparation of free CPT and TPT stock solution
8.7 mg of CPT powder or 10.5 mg of TPT powder were dissolved in 25 mL of DMSO in a volumetric flask. The final concentrations were 10−3 M. They were covered with aluminum foil and stored at 4 °C in the refrigerator. They were brought to room temperature and kept approx. 1 h for the dissolution of solvent prior to use in the analysis.Preparation of triethyl amine acetate (TEAA) buffer
10 mL of triethyl amine was added to 990 mL of distilled water in a volumetric flask. The pH of the system was adjusted to 5.5 with glacial acetic acid. Suction filtration was performed for the removal of potential dusts, and it was degassed prior to use.Fluorescence spectroscopy analysis
5 μL of drug loaded gel was diluted with approx. 3 mL of pH = 7.4 tris buffer in a quartz cell. Then, it was immediately analyzed with fluorescence spectroscopy using a double wavelength program. The wavelengths (λex, λ1 em, λ2 em) employed are shown in Table 4. For the control experiments; 3 μL of 10−3 M CPT in DMSO was diluted with 3 mL pH = 7.4 tris buffer in a quartz cell. In addition, 5 μL of 10−3 M TPT in DMSO was diluted with 3 mL pH = 7.4 tris buffer in a quartz cell.| ID | λ excitation (nm) | λ1 emission (nm) | λ2 emission (nm) |
|---|---|---|---|
| Free CPT | 370 | 432 | 480 |
| Free TPT | 381 | 516 | 580 |
| CPT in gel | 370 | 432 | 480 |
| TPT in gel | 381 | 516 | 580 |
Scatchard plots were used to evaluate the [L]/[C] ratio.20 Concentrations of the ring closed (lactone) and opened form (carboxylate) of drugs were referred to as [L] and [C], respectively.
A fluorescence emission intensity I(λ1) of a sample consisting of both lactone and carboxylate forms at a given time can be described as
| I(λ1) = Ic(λ1) * [C] + IL(λ1) * [L] (eqn 1) |
Similarly for I(λ2),
| I(λ2) = Ic(λ2) * [C] + IL(λ2) * [L] (eqn 2) |
The ratio of the fluorescence intensities of the sample at λ1 and λ2 wavelengths gives the R factor.
| R = I(λ1)/I(λ2) (eqn 3) |
Then, [C]/[L] ratio can be simply written as
| [C]/[L] = [IL(λ2)/Ic(λ2)] * (RL-R)/(R-Rc) (eqn 4) |
Lactone fraction [L%] in the sample can be calculated as
| [L%] = 100%/{1 + [IL(λ2)/Ic(λ2)] * (RL-R)/(R-Rc)} (eqn 5). |
Scatchard plots of hydrolysis can be plotted as the lactone percentage [L%] or carboxylate percentage [C%] with respect to time (t). Each point of plot is the result of an average of at least three independent reproductions.
Analysis of RL and RC values for free CPT and TPT via fluorescence spectroscopy
RL and RC values are critical factors for the estimation of the lactone percentage [L%]. Fluorescence spectroscopy was employed to measure the intensities of free camptothecin and topotecan in DMSO, and further diluted with tris buffer at either acidic or basic pH conditions. Intensities were recorded at two different emission wavelengths, and then RL and RC were found by calculating the ratio of the measured intensities.Analysis of RL and RC values for CPT and TPT in gel via fluorescence spectroscopy
A similar methodology as above was also used for the analysis of RL and RC of drug loaded gels. Intensities of pure carboxylate and lactone forms of CPT and TPT at different emission wavelengths in copolymer gel were measured at both acidic and basic pH conditions. Then, the RL and RC values for CPT and TPT in gel were calculated.HPLC analysis
A simple and versatile high-performance liquid chromatographic method was used to separate the lactone and carboxylate forms of free CPT and TPT drugs.21 This two component mobile phase system contains triethylamine acetate (TEAA) and acetonitrile (ACN). In this method, triethyl amine has multiple advantages. It is used as an ion pairing reagent, a masking agent for underivatized silanols, and a major buffer constituent for the pH adjustment. Thus, it provides sufficient retention and good separation for carboxylate species on the column. On the other hand, ACN was employed to control the lactone species of drugs.5 μL of the drug loaded gel was diluted with approx. 1 mL of pH = 7.4 tris buffer in a vial. Then, it was immediately analyzed with HPLC having a C18 reverse phase column and fluorescence detector. Excitation (λex) and emission (λ1 em) wavelengths used in HPLC analysis were shown in Table 4. In addition, the mobile phase that was used was 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :77 (v/v) ACN-TEAA buffer (1%, v/v, pH: 5.5) for CPT analysis, and 12:88 (v/v) ACN-TEAA buffer (2%, v/v, pH: 5.5) for TPT analysis.21 For control experiments; 3 μL of 10−3 M CPT in DMSO was diluted with 1 mL pH = 7.4 tris buffer in vial. In addition, 5 μL of 10−3 M TPT in DMSO was diluted with 1 mL pH = 7.4 tris buffer in vial. Then, it was immediately analyzed with HPLC equipped with a fluorescence detector using the wavelengths shown in Table 4.
:77 (v/v) ACN-TEAA buffer (1%, v/v, pH: 5.5) for CPT analysis, and 12:88 (v/v) ACN-TEAA buffer (2%, v/v, pH: 5.5) for TPT analysis.21 For control experiments; 3 μL of 10−3 M CPT in DMSO was diluted with 1 mL pH = 7.4 tris buffer in vial. In addition, 5 μL of 10−3 M TPT in DMSO was diluted with 1 mL pH = 7.4 tris buffer in vial. Then, it was immediately analyzed with HPLC equipped with a fluorescence detector using the wavelengths shown in Table 4.
Analysis of the lactone conversion of free CPT and TPT via HPLC
Retention time of pure carboxylate and pure lactone of free CPT and TPT was determined at a basic pH (9.9) and acidic pH (5.0), respectively. HPLC analysis was performed with a C-18 column using the ACN- TEAA buffer mobile phase. The spectral data was given in the section of “copy of HPLC spectra” in the ESI.†Analysis of the lactone conversion of CPT and TPT in gel via HPLC
A similar methodology as described above was used to test which form of drugs exists in gel at pH = 7.4 over time. Firstly, the 0.015% loading of CPT and TPT into gel was examined with HPLC. We also analyzed higher drug loadings, such as 1% and 10%. Spectral data was given in the section of “copy of HPLC spectra” in the ESI.†Release experiments
Topotecan and camptothecin were loaded into copolymer gel with 0.015% and 1.0% loading ratios (Table 5). For example, 0.0375 mg TPT and 250 mg polymer were mixed homogeneously in an Eppendorf tube. 350 μL of pH = 7.4 tris buffer was added to form a gel at room temperature. 350 μL of buffer at pH = 7.4 was suitable to keep the matrix in a gel form at 35 °C. 0.5 mL of pH = 5 tris buffer, which inhibits the fluctuation between carboxylate and lactone intensities, was added into the upper side. Therefore, the system contains two layers; the first layer is a gel with a drug while the second layer is a supernatant. Other formulations were prepared in the same manner as indicated in Table 5. Then, they were put into a shaker having a constant speed at 160 rpm and 35 °C. At different time points, the supernatants were replaced with fresh ones. Measurements were carried out using fluorescence spectroscopy with single read measurements at the appropriate wavelengths of drugs as shown in Table 4. Dilutions were performed prior to the measurements, if necessary. Furthermore, a blank formulation, having no drug, was prepared for the elimination of the low fluorescence characteristic of the polymer.Examination of CPT loaded gel with ATR-FTIR
For the stabilization mechanism, the ATR-FTIR spectra of powder CPT and CPT loaded copolymer gel were recorded, respectively. For the CPT loaded gel sample, a highly concentrated copolymer solution (almost like gel) was prepared with PLLA-mPEG and distilled water. It was recorded as background. Then, 1–2 mg of powder CPT was added into the same vial before it was stirred with a high speed vortex for the complete homogenization. The ATR-FTIR spectrum of the drug loaded gel was recorded, and the background was subtracted. Thus, the ATR-FTIR spectra of powder CPT (1) was compared to the CPT loaded copolymer gel (2).Cell culture
4T1 mouse mammary carcinoma cell line (American Type Culture Collection, ATCC; Manassas, VA, USA) and mouse Lewis lung carcinoma (LLC-1) cells were cultured in 75 cm2 culture flasks containing RPMI1640 culture media supplemented with heat inactivated 10% fetal bovine serum, 2 mM L-glutamine (Thermo Scientific, HyClone, Logan, UT, USA), 100 units mL−1 penicillin and 100 μg mL−1 streptomycin (Biochrom, Berlin, Germany) at 37 °C in a humidified incubator containing 5% CO2.MTT assay
Topotecan (TPT) was dissolved in DMSO (80 mg mL−1) as a stock solution, and test concentrations were prepared freshly in culture media. TPT in the PLLA-mPEG platform (0.25% loading) overlaid by culture media was allowed for the release of the drug for two hours. Then, the culture media was collected and test dilutions were prepared for the MTT assay. The same procedure was performed with PLLA-mPEG gel as a negative control. The amount of released TPT in the culture media was fluorometrically quantitated prior to the cytotoxicity assay.The methlythiazolyltetrazolium (MTT) assay was used to evaluate cell viability.22 Briefly, 50 μL cell suspensions containing 3 × 104 or 1 × 104 4T1 and 1 × 104 LLC-1 cells were seeded in 96-well flat-bottom plates, and varying concentrations of freshly dissolved TPT or TPT released from the PLLA-mPEG gel were added into each well (50 μL/well). All concentrations were studied in quadruplicates. Then, the cells were kept for a period of 24 h at 37 °C in a humidified incubator containing 5% CO2. After 24 h of incubation, 25 μL of MTT solution (5 mg mL−1; Sigma, St. Louis, MO, USA) were added to each well, and the plates were incubated for a further 4 h. The formazan precipitate was solubilized by adding 80 μL lysing buffer (pH = 4.7) composed of 23% SDS (Sigma) dissolved in 45% N,N-dimethylformamide solution (Sigma). After an overnight incubation at 37 °C, the optical densities (OD) were read at 570 nm using a microplate reader (Spectramax Plus, Molecular Devices, Sunnyvale, CA, USA). Cells incubated in the culture medium alone served as a control for cell viability (untreated wells). Cell viability (%) was calculated as (OD of treated wells/OD of untreated wells) ×100.
Animals and tumor formation
Six- to eight-week-old inbred female BALB/c mice (Kobay As., Ankara, Turkey) were housed under environmentally controlled standard conditions. The Guiding Principles in the Care and Use of Laboratory Animals together with those described in the Declaration of Helsinki were strictly adhered to in the conduct of all the experimental procedures described within this manuscript. This project was approved by the Institutional Ethical Committee of Hacettepe University, Ankara, Turkey (Approval Number: 2010/10-10) before its commencement.4T1 mouse mammary carcinoma cells at the fourth passage (5 × 104 cells suspended in 0.1 mL phosphate-buffered saline) were inoculated subcutaneously into the right dorsal flank of the syngeneic BALB/c mice.15 The mice were distributed into four experimental groups: Control group (injected with physiological saline, 0.9% NaCl, 0.13 mL/30 gr; n = 7), PLLA-mPEG control group (injected with PLLA-mPEG gel, 0.13 mL/30 gr; n = 10), TPT group (injected with Topotecan dissolved in physiological saline (0.35 mg mL−1; n = 10), 0.045 mg TPT/kg), and PLLA-mPEG-TPT group (injected with PLLA-mPEG-TPT gel (0.35 mg mL−1 (0.08% TPT loading), 0.045 mg TPT/kg; n = 10). Peri-tumoral subcutaneous injections were performed during the second week of post-inoculation coinciding with the appearance of palpable tumors. The size of the tumors were measured twice a week, and the mean diameter was calculated as the square root of the product of the two perpendicular diameters.
Statistical analysis
All the values are expressed by arithmetic mean ± standard deviation (SD). The statistical difference between the experimental groups was determined using a Student's paired t-test where appropriate. The differences were regarded as statistically significant when P ≤ 0.05.Results and discussion
Syntheses and sol–gel properties of mPEG-PLLA diblock copolymers
The synthesis of injectable biodegradable poly(L-lactic acid)-methoxy poly(ethylene glycol) diblock copolymers (PLLA-mPEG) was performed as shown in Fig. 2.19 mPEG (2000 g mol−1) was used to synthesize copolymer because the higher molecular weight of PEG (above ∼10000) is inappropriate for filtration through a human kidney membrane due to the large hydrodynamic radius of the PEG in the aqueous phase.1 In addition, the hydrophobic PLLA length was adjusted by the changing amount of L-lactide at a constant mass of hydrophilic mPEG, as indicated in the experimental section (Tables 1 and 2).
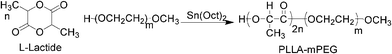 | ||
| Fig. 2 PLLA-mPEG polymerization starting from L-lactide and mPEG. | ||
PLLA-mPEG copolymers, bearing an appropriate length of each block component, shows sol behavior at around 45 °C, which is suitable for the injection, and then a gel with subsequent rapid cooling to body temperature.1 First, copolymers 1–3 were used for the examination of the potential sol–gel properties. Copolymers 1 and 2 were not homogeneously suspended at low concentration (3%) because of higher lactide content relative to the mPEG moiety in a diblock chain. On the other hand, copolymer 3 was homogeneously suspended, and formed a sol property at room temperature at 3, 5, 10, and 30% (mg μL−1). Then, a 45% polymer suspension of 3 was tested to examine gel to sol temperature. While it was gel at rt, it started to flow immediately at 35–37 °C (Table 6). Then, TPT and CPT anticancer drugs were loaded into gels, respectively. The homogeneous dispersion of drugs within gels was obtained for all the selected ratios (0.015, 1.0, and 10%).
| ID (copolymers) | 1 | 2 | 3 | |
|---|---|---|---|---|
| a N.O.: Not Observed; r.t.: room temp | ||||
| Sol–gel temp. °C | N.O. | N.O. | <r.t. | 37 |
| Conc% (mg μL−1) | 3 | 3 | 3, 5, 10, 30 | 45 |
Stability of CPT and TPT in gels
This is the first time the combination of a polymer-TPT (or CPT) in gel was examined with fluorescence spectroscopy (FL) to elucidate the kinetics of lactone hydrolysis by exploiting the methodology for the analysis of free camptothecin series via fluorescence spectroscopy.20 The behaviors of CPT and TPT in gels at pH = 7.4 together with the controls (drugs in supernatants) were evaluated for the ratio of fluorescence intensities of two different emission wavelengths as a function of time. The ratios of intensities at two different emissions of wavelength of pure lactone (RL) and pure carboxylate (RC) were also measured for each species and thereafter employed as constants (Table 7). Then, ring closed (lactone) [L]% and an opened form (carboxylate) [C]% of drugs were calculated by scatchard plots and were found by| [L%] = 100%/{1 + [IL(λ2)/Ic(λ2)] * (RL-R)/(R-Rc)} |
| ID | RL | RC |
|---|---|---|
| Free CPT | 3.43 | 2.18 |
| Free TPT | 4.86 | 5.41 |
| CPT in gel | 2.39 | 2.07 |
| TPT in gel | 2.89 | 3.70 |
The kinetics of the lactone hydrolysis of 0.015% CPT and TPT loaded gels performed with fluorescence spectroscopy at pH = 7.4 showed that >95% of CPT and TPT maintained its lactone form in gel during approx. 5 h (Fig. 3c and d, see Tables 1–4 in the ESI† for detailed measurements and calculations).
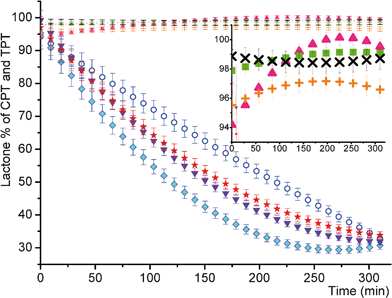 | ||
| Fig. 3 Stability of CPT and TPT in gels (0.015%) versus rapid hydrolysis in physiological environment (a. circle: L% of free TPT in buffer analyzed with FL; b. diamond: L% of free CPT in buffer analyzed with FL; c. up-triangle: L% of CPT in gel analyzed with FL; d. plus: L% of TPT in gel analyzed with FL; e. down-triangle: L% of free CPT in buffer analyzed with HPLC; f. star: L% of free TPT in buffer analyzed with HPLC; g. cross: L% of TPT in gel analyzed with HPLC; h. square: L% of CPT in gel analyzed with HPLC). Each point of the plot is the result of an average of at least three independent reproductions. Polynomial fitting of the data was performed. | ||
The kinetics of the lactone hydrolysis of CPT and TPT in gels was also monitored by high-performance liquid chromatography by exploiting the methodology for the analysis of free camptothecin series.21 The retention time of pure carboxylate and pure lactone of free CPT and TPT was determined at basic pH (9.9) and acidic pH (5.0), respectively. The carboxylate peaks of CPT and TPT appeared at 1.51 and 1.53 min while lactone peaks gave signals at 4.21 and 3.72 min. When the kinetic behaviors of CPT and TPT in tris buffer at pH = 7.4 were examined with time elapsed, a decrease in lactone form was observed in Fig. 3e and f (representative spectra were shown at Fig. 1 and 2 in the ESI†). A similar methodology above was used to test which form of drugs exists in gel at pH = 7.4 over time. Firstly, the 0.015% loading of CPT and TPT into gel was examined with HPLC. It was observed that each drug maintained its lactone form (>98%) in gel over time (Fig. 3g and h). Moreover, higher drug loadings such as 1.0% and 10% showed a high stability of lactone species (>98%), as indicated in Table 8.
| CPT in gel, L (%) | TPT in gel, L (%) | |||
|---|---|---|---|---|
| Time (min) | 1% loading | 10% loading | 1% loading | 10% loading |
| 0 | 99.9 | 100 | 99.1 | 98.6 |
| 30 | 99.9 | 99.8 | 99.1 | 98.1 |
| 60 | 99.9 | 99.8 | 99.2 | 98.4 |
| 90 | 99.9 | 99.9 | 98.8 | 98.4 |
| 120 | 99.9 | 99.8 | 98.9 | 98.7 |
| 150 | 99.9 | 99.9 | 98.8 | 98.6 |
| 180 | 99.9 | 99.8 | 97.8 | 98.8 |
Concentration effect on drug stability
It was also examined as to how CPT and TPT behaved at concentrated PLLA-mPEG solutions (∼250 mg mL−1) at room temperature. Surprisingly, HPLC measurements revealed that CPT and TPT still keep lactone intact. Finally, CPT and TPT were tested in very aqueous polymer solutions (500× dilutions from gel). This formulation slowed the hydrolysis of drugs at pH = 7.4 in buffer (almost a 20% increase in the stability when compared to free drugs at pH = 7.4 buffer). Thus, with the bulk or surface erosion of a biodegradable polymer in the tumor environment, the continuation of drug interaction with the surface eroded polymer in the tumor environment may trigger slow drug degradation even after the drug is released. This may enhance the efficiency of therapy.Release CPT and TPT from gel
Fig. 4 shows the release profile of CPT and TPT from gels. Each point represents the amount of drugs left from the gels. There was a constant daily release of 0.015% loadings of both drugs without any significant burst effect (only 5 to 10% burst release in a couple of hours). Similar behaviors were observed for 1.0% loadings as well. After 23 days, 75% and 89% of the total CPT and TPT mass were released from the gels with 0.015% loadings of both drugs, respectively. TPT has a hydrophilic character and is relatively more soluble in water, while hydrophobic CPT has low water solubility. TPT release is faster than CPT. Thus, TPT moves fast to the upper buffer layer. In addition, the complete release of the drugs from the gels was observed for 1.0% loadings of both drugs at the end of 23 days (Fig. 4).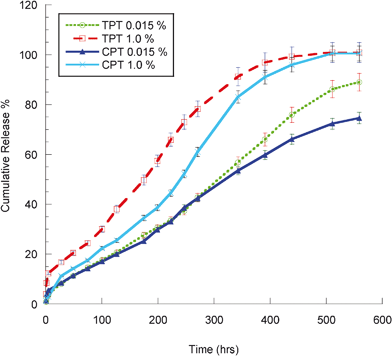 | ||
| Fig. 4 CPT and TPT release curves | ||
Mechanistic outlook on the stabilization of CPT via PLLA-mPEG gels
The mechanistic outlook on the stabilization of CPT via PLLA-mPEG gels was also mechanistically examined in detail. The 20–OH group of the lactone ring at camptothecin plays an important role in the ring opening of the lactone moiety of the drug.10–15,24–26 Proton transfer or the stabilization of the transition state via a strong intramolecular hydrogen bonding may facilitate the ring opening of the ring after a nucleophilic attack occurred at the acyl carbon10–15 (Fig. 5). It was also reported that the esterification of the 20–OH of CPT stabilized the lactone ring, supporting this phenomenology.26 Intermolecular interactions between the 20–OH group of the lactone moiety and carbonyl groups of the lactide units in PLLA-mPEG diblock copolymer most probably reduce the intramolecular hydrogen bonding within the lactone moiety of a drug molecule. ATR-FTIR might be a good aspect to elucidate those interactions. Thus, the ATR-FTIR spectrum of powder CPT (1) was compared to the CPT loaded gel (2) (Fig. 3 in the ESI†). It was observed that three bands in the spectra located at 3100–3500, 1360–1390, and 650–720 cm−1 had undergone significant changes and should be closely analyzed. The band around 3427 cm−1 was assigned to the O–H stretching vibration of CPT. This bond highly shifted to 3658 cm−1 due to potential intermolecular hydrogen bonding between the carbonyl group of the copolymer and the hydroxyl group of CPT. While the O–H in-plane bending vibration of powder CPT was observed at 1387 cm−1 with a weak small signal, that of the drug loaded copolymer gel was observed at 1390 cm−1 with a strong broad signal. In addition, the spectral changes 650–720 cm−1 might have resulted from the out-of-plane bending of the O–H group.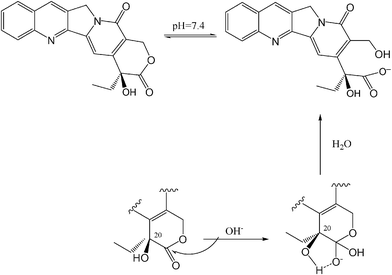 | ||
| Fig. 5 Proposed mechanism of the conversion of the lactone ring to the carboxylate form of CPT.10 | ||
In addition, as expected, there was no shift in the carbonyl stretching of the lactone ring (1737 cm−1), lactame (1650 cm−1) ring, and aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (1578 cm−1 and 1600 cm−1) of powder CPT and CPT loaded copolymer gel. It is known that carboxylate ion gives rise to two bands: the asymmetrical stretching band near 1650–1550 and a weaker, symmetrical stretching band near 1400 cm−1. These bands did not appear in the spectrum of the drug loaded copolymer gel, which was additional proof of the active lactone ring of CPT in gel (Fig. 3 in the ESI†).
C bonds (1578 cm−1 and 1600 cm−1) of powder CPT and CPT loaded copolymer gel. It is known that carboxylate ion gives rise to two bands: the asymmetrical stretching band near 1650–1550 and a weaker, symmetrical stretching band near 1400 cm−1. These bands did not appear in the spectrum of the drug loaded copolymer gel, which was additional proof of the active lactone ring of CPT in gel (Fig. 3 in the ESI†).
Confocal and bright field microscopes were used to examine the characteristics of some CPT crystals that might give us a complementary idea about the mechanism for CPT stabilization. Free CPT suspended water was previously reported by SEM27 and had the image of the lamellar square type of crystals due to the low solubility of CPT crystals in water. When the CPT molecules in gel were examined by a confocal and optic microscope, the similar behavior of CPT molecules was observed as shown in Fig. 6A–B. The conversion of the lactone ring to water soluble carboxylate form in a gel state is quite slow due to potential intermolecular interactions via hydrogen bonding between the 20S group and carbonyl group of the lactide units. Those strong interactions may inhibit the formation of the water soluble carboxylate form.
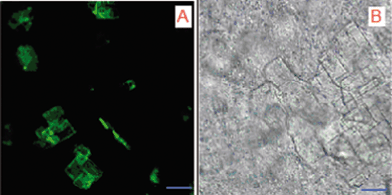 | ||
| Fig. 6 Confocal (A) and bright field (B) microscope images of drug loaded gels. Scale was 10 μm. | ||
In vitro studies
The cytotoxic efficacy of TPT in the PLLA-mPEG platform (PLLA-mPEG-TPT) was evaluated on LLC-1 and 4T1 cancer cell lines by MTT assay. The amount of TPT released into the media ranged between 363–402 μM in independent experiments. TPT released from the gel exerted cytotoxic effects on LLC-1 tumor cells (Fig. 7). 4T1 cells were sensitive neither to released TPT nor to fresh TPT solution (Fig. 8A). Notably, a very high dose of TPT (200 μM) decreased 4T1 viability (Fig. 8B). The effect of TPT on cell survival can be related to the malignant character and the origin of tumors.28 In a widely used human breast cancer cell line, MDA-MB-231, sensitivity to TPT was also observed at high drug concentrations.28 4T1 mouse mammary carcinoma cells are also aggressive and chemoresistant.23 Therefore, the insensitivity of 4T1 cells to TPT may be attributed to drug resistance besides for the short-time drug exposure, in vitro.23,28 However, because of the highly malignant nature of 4T1, this cell line is regarded as a compatible model for studying human breast cancer.23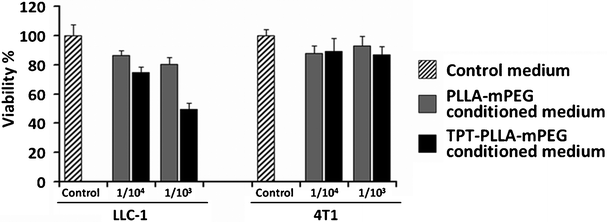 | ||
| Fig. 7 Effect of TPT released from PLLA-mPEG gels on the viability of LLC-1 and 4T1 cancer cells. 1 × 104 cells were seeded and 1:104 and 1:103 dilutions of the conditioned media were tested. | ||
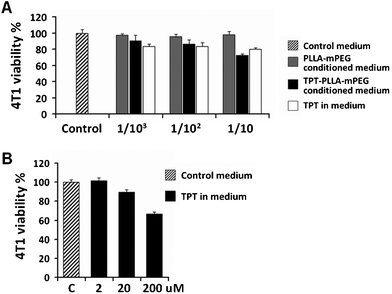 | ||
| Fig. 8 Very high doses of TPT decreased the viability of 4T1 cancer cells. (A) Effect of TPT released from PLLA-mPEG gel, TPT solution, and PLLA-mPEG conditioned cell culture media on the 4T1 cell viability. 3 × 104 cells were seeded and 1:103, 1:102, and 1:10 dilutions (0.4 μM, 4 μM and 40 μM, respectively) of TPT solution or released drug were tested. (B) Dose response for the TPT solution was also determined. A statistical difference was obtained between the control cells and the cells treated with 200 μM TPT. | ||
In vivo studies
Tumors established with 4T1 cells are widely accepted as a superior model compatible with human breast tumors23 and we challenged the efficacy of the TPT-loaded PLLA-mPEG preparation on this model. When compared to the positive and negative control groups, the administration of PLLA-mPEG-TPT to the tumor-bearing mice resulted in a significant reduction in tumor size (Fig. 9A and B). The initial reduction (day 3) seen with the PLLA-mPEG-TPT was compatible with the TPT solution. However, on the seventh day, the tumor size in the group treated with TPT solution reached that of the control group, whereas PLLA-mPEG-TPT considerably hampered the tumor growth during 14 days of follow-up (Fig. 9A and B). This result simply demonstrates the increased in vivo stability of TPT when administered in PLLA-mPEG gel. Correspondingly, between the days 14–17, PLLA-mPEG-TPT gel improved the survival rate of mice bearing tumors established with 4T1 cells, which were insensitive to TPT, in vitro (Fig. 9C). In the study of Berrada et al.,18 in demonstrating the treatment of potentially drug-sensitive tumors, the biodegradable polymer gel-CPT preparation was administered directly into the tumor mass, but its efficacy was compared with the CPT solution that was given intraperitoneally.18 In our study, the usage of TPT-insensitive 4T1 cells was the major challenge in our study to test the efficacy of TPT-PLLA-mPEG. In addition, all the agents (control physiological saline, TPT solution, PLLA-mPEG gel, and TPT-PLLA-mPEG) were administered via the same route.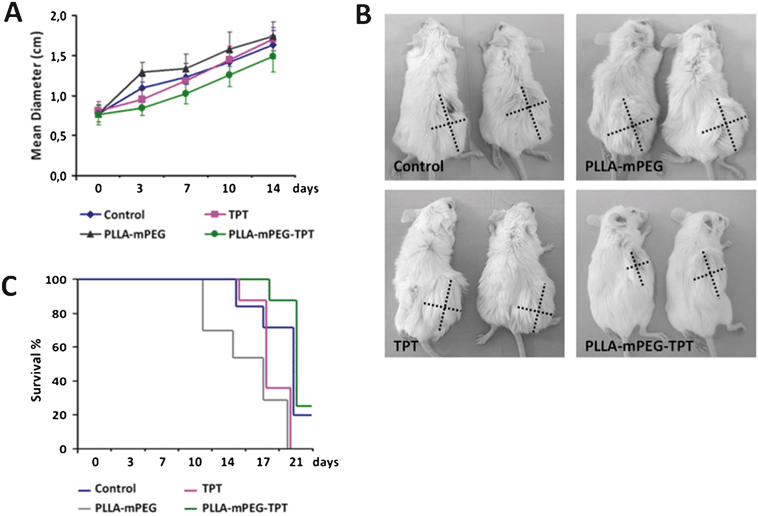 | ||
| Fig. 9 (A) The change in 4T1 breast tumors' sizes during 14 day follow-up in groups treated with control saline, TPT solution, PLLA-mPEG, and TPT-loaded PLLA-mPEG. (B) Representative photographs of two mice bearing 4T1 tumors from each experimental group are shown. The dashed lines indicate the perpendicular diameters of the tumors. (C) Survival percentages of the mice bearing 4T1 breast tumors during the treatment period can be seen. | ||
Conclusions
The synthesis of biodegradable PLLA homopolymers and PLLA-mPEG diblock copolymers for the formation of thermo-sensitive gels were performed. PLLA-mPEG copolymers, having an appropriate length of each block component, showed sols at around 45 °C, suitable for the injection, then a gel with subsequent rapid cooling to body temperature. Topotecan and camptothecin were selected as anticancer drugs. Both drugs can easily hydrolyze under physiological conditions (pH = 7.4). This causes the loss of its activity and turns into the inactive toxic carboxylate form from the active lactone form. To keep those anticancer drugs in the lactone form, they were loaded into PLLA-mPEG gels in different loading ratios. Their stability in gel was examined with HPLC and fluorescence spectroscopy. It was found that both drugs were highly stable in the prepared gels (>95%). Then, both release profiles of the drugs showed a prolonged release over the course of weeks. Mechanistic studies on the stabilization of the CPT anticancer drug with PLLA-mPEG gels were carried out using ATR-FTIR, confocal and optic microscopes. The cytotoxic efficacy of TPT in the PLLA-mPEG platform (PLLA-mPEG-TPT) was evaluated on the LLC-1 and 4T1 cancer cell lines by MTT assay. In vivo administration of PLLA-mPEG-TPT to the mice with breast tumors established with 4T1 cells resulted in a reduction in tumor size and better survival percentages in mice. In addition, the stabilization of CPT and TPT with gels may find another application on solid tumors in the brain via local injection.Acknowledgements
We would like to thank Mr Yusuf Dolen. The financial support from the Scientific and Technological Research Council of Turkey (TÜBITAK), Turkish Academy of Sciences (TÜBA), and the Middle East Technical University (METU) are all gratefully acknowledged.References
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Biodegradable block copolymers as injectable drug-delivery systems, Nature, 1997, 388(6645), 860–862 CrossRef CAS.
- I.C. Kwon, Y.H. Bae and S.W. Kim, Electrically erodible polymer gel for controlled release of drugs, Nature, 1991, 354, 291–293 CrossRef CAS.
- R. Langer, New Methods of Drug Delivery, Science, 1990, 249, 1527–1533 CAS.
- G. Chandrashekar and N. Udupa, Biodegradable injectable implant systems for long term drug delivery using poly (lactic-co-glycolic) acid copolymers, J. Pharm. Pharmacol., 1996, 48(7), 669–74 CrossRef CAS.
- C. I. Winternitz, J. K. Jackson, A. M. Oktaba and H. M. Burt, Development of a polymeric surgical paste formulation for taxol, Pharm. Res., 1996, 13(3), 368–75 CrossRef CAS.
- M. Katakam, W.R. Ravis and A.K. Banga, Controlled release of human growth hormone in rats following parenteral administration of poloxamer gels, J. Controlled Release, 1997, 49, 21–26 CrossRef CAS.
- A. Bajpai, S. Shukla, R. Saini and A. Tiwari, Stimuli Responsive Drug Delivery Systems - From Introduction to Application, Smithers Rapra Technology, 2010 Search PubMed.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Polymeric Vesicles: From Drug Carriers to Nanoreactors and Artificial Organelles, Acc. Chem. Res., 2011, 44(10), 1039–1049 CrossRef CAS.
- C. Ferraro, L. Quemeneur, S. Fournel, A. F. Prigent, J. P. Revillard and N. Bonnefoy-Berard, The topoisomerase inhibitors camptothecin and etoposide induce a CD95-independent apoptosis of activated peripheral lymphocytes, Cell Death Differ., 2000, 7(2), 197–206 CrossRef CAS.
- J. Fassberg and V. J. Stella, A Kinetic and Mechanistic Study of the Hydrolysis of Camptothecin and Some Analogs, J. Pharm. Sci., 1992, 81(7), 676–684 CrossRef CAS.
- M. C. Wani, A. W. Nicholas, G. Manikumar and M. E. Wall, Plant Antitumor Agents .25. Total Synthesis and Antileukemic Activity of Ring a Substituted Camptothecin Analogs - Structure Activity Correlations, J. Med. Chem., 1987, 30(10), 1774–1779 CrossRef CAS.
- M. C. Wani, A. W. Nicholas and M. E. Wall, Plant Antitumor Agents .28. Resolution of a Key Tricyclic Synthon, 5′(Rs)-1,5-Dioxo-5′-Ethyl-5′-Hydroxy-2′h,5′h,6′h-6′-Oxopyrano[3′,4′-F]Delta-6,8-Tetrahydroindolizine - Total Synthesis and Antitumor-Activity of 20(S)-Camptothecin and 20(R)-Camptothecin, J. Med. Chem., 1987, 30(12), 2317–2319 CrossRef CAS.
- C. Jaxel, K. W. Kohn, M. C. Wani, M. E. Wall and Y. Pommier, Structure–Activity Study of the Actions of Camptothecin Derivatives on Mammalian Topoisomerase-I - Evidence for a Specific Receptor-Site and a Relation to Antitumor-Activity, Cancer Res., 1989, 49(6), 1465–1469 CAS.
- T. G. Burke, A. S. Demir, C. Tanyeli, A. J. Chavan, T. L. Wang, Y. Pommier, Water-soluble derivatives of camptothecin/homocamptothecin, US Pat., 2001, 6,291,676.
- T. G. Burke, A. S. Demir, A. J. Chavan, D. Yang, Oligonucleotide delivery systems for camptothecins, US Pat., 2005, 6,897,200.
- J. M. Gao, J. Ming, B. He, Z. W. Gu and X. D. Zhang, Controlled release of 9-nitro-20(S)camptothecin from methoxy poly(ethylene glycol)-poly(D,L-lactide) micelles, Biomed. Mater., 2008, 3, 15013 CrossRef CAS.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, Injectable block copolymer hydrogels for sustained release of a PEGylated drug, Int. J. Pharm., 2008, 348(1–2), 95–106 CrossRef CAS.
- M. Berrada, A. Serreqi, F. Dabbarh, A. Owusu, A. Gupta and S. Lehnert, A novel non-toxic camptothecin formulation for cancer chemotherapy, Biomaterials, 2005, 26(14), 2115–2120 CrossRef CAS.
- O. Mert, E. Doganci, H. Y. Erbil and A. S. Dernir, Surface characterization of poly(L-lactic acid)-methoxy poly(ethylene glycol) diblock copolymers by static and dynamic contact angle measurements, FTIR, and ATR-FTIR, Langmuir, 2008, 24(3), 749–757 CrossRef CAS.
- I. Chourpa, J. M. Millot, G. D. Sockalingum, J. F. Riou and M. Manfait, Kinetics of lactone hydrolysis in antitumor drugs of camptothecin series as studied by fluorescence spectroscopy, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379(3), 353–366 CrossRef CAS.
- D. L. Warner and T. G. Burke, Simple and versatile high-performance liquid chromatographic method for the simultaneous quantitation of the lactone and carboxylate forms of camptothecin anticancer drugs, J. Chromatogr., Biomed. Appl., 1997, 691(1), 161–171 CrossRef CAS.
- M. B. Hansen, S. E. Nielsen and K. Berg, Re-Examination and Further Development of a Precise and Rapid Dye Method for Measuring Cell-Growth Cell Kill, J. Immunol. Methods, 1989, 119(2), 203–210 CrossRef CAS.
- B. A. Pulaski and S. Ostrand-Rosenberg, Mouse 4T1 breast tumor model, Curr. Protoc. Immunol. 2001, ch. 20, unit 20.2 Search PubMed.
- M. E. Wall, Alkaloids with antitumor activity. In International Symposium on Biochemistry and Physiology of the Alkaloids, ed. K. Mothes, K. Schreiber and H. R. Schutte, Academie-Verlag, Berlin, 1969, pp. 77–87 Search PubMed.
- A. W. Nicholas, M. C. Wani, G. Manikumar, M. E. Wall, K. W. Kohn and Y. Pommier, Plant Antitumor Agents .29. Synthesis and Biological-Activity of Ring-D and Ring-E Modified Analogs of Camptothecin. Journal of Medicinal Chemistry, 1990, 33(3), 972–978 CAS.
- H. Zhao, C. Lee, P. K. Sai, Y. H. Choe, M. Boro, A. Pendri, S. Y. Guan and R. B. Greenwald, 20-O-acylcamptothecin derivatives: Evidence for lactone stabilization, J. Org. Chem., 2000, 65(15), 4601–4606 CrossRef CAS.
- V. Kumar, J. C. Kang and R. J. Hohl, Improved dissolution and cytotoxicity of camptothecin incorporated into oxidized-cellulose microspheres prepared by spray drying, Pharm. Dev. Technol., 2001, 6(3), 459–467 CrossRef CAS.
- S. S. Lin, L. Sun, Y. K. Zhang, R. P. Zhao, W. L. Liang, S. T. Yuan and L. Y. Zhang, Topotecan inhibits cancer cell migration by down-regulation of chemokine CC motif receptor 7 and matrix metalloproteinases, Acta Pharmacol. Sin., 2009, 30(5), 628–636 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of some experimental calculations and measurements, and spectral data, are included in this section. See DOI: 10.1039/c1ra00366f |
| ‡ On leave of absence from Kocaeli University, 41380, Kocaeli, Turkey. |
| This journal is © The Royal Society of Chemistry 2012 |