DOI:
10.1039/C1RA00417D
(Paper)
RSC Adv., 2012,
2, 669-676
Characteristics of the charge transfer surface complex on titanium(IV) dioxide for the visible light induced chemo-selective oxidation of benzyl alcohol†
Received
7th July 2011
, Accepted 2nd October 2011
First published on 17th November 2011
Abstract
This paper deals with the oxidation of benzyl alcohol by O2 on pure TiO2 under visible light irradiation, and it was found that the benzyl alcohol is converted into benzaldehyde with high selectivity (>99%). In order to understand the origins of the visible light induced photocatalysis, surface characterizations of the charge transfer complex formed by the interaction of benzyl alcohol with the TiO2 was extensively performed by investigating the effect of heat-treatment on TiO2 or its chemical modification with hydrofluoric acid. Moreover, the study of the kinetic isotope effect (KIE) for the oxidation of benzyl alcohol showed that the α-deprotonation from benzyl alcohol is the rate determining step (RDS), the process of which is assisted by the terminal OH groups of TiO2. Photo-electrochemical investigations were also incorporated to demonstrate the reaction mechanism behind the visible light induced photocatalytic reaction.
Introduction
The selective oxidation of alcohols into carbonyl compounds is a fundamental and significant procedure with great potential for scientific and industrial applications. In particular, the use of clean oxidants such as O2 is highly desired for improving reaction atom efficiency, and an increasing need to avoid conventional stoichiometric oxidizing agents such as Cr (+VI), peroxy acids and basic media.
Titanium(IV) dioxide (TiO2) is one of the most extensively investigated materials in the challenge to address such energy and environmental needs1–6 as H2 production by water splitting, the degradation of volatile organic compounds (VOC), photocatalytic organic syntheses, dye-sensitized solar cells, and super-hydrophilic materials. Among them, chemo-selective organic syntheses over TiO2 photocatalysts have been the focus of much attention.6–29 In particular, the selective oxidation of alcohol such as benzylic alcohol into corresponding carbonyl compounds by O2 were studied in an organic solvent,16–20 in gas phase,21 or in aqueous solution22–29 by band-gap excitation (hv > 3.2 eV) of TiO2. However, in order to take advantage of the unlimited solar light available, photocatalytic systems which work under visible light (400–800 nm) irradiation is strongly desired. Very recently, Tada et al. demonstrated the plasmon-induced selective alcohol oxidation in water over gold nano-particles supported TiO2 (Au/TiO2) photocatalyst under visible light irradiation.30 It was also reported that visible light illumination of nitrogen-doped TiO2 brings about the selective oxidation of benzyl and cinnamyl alcohol to the corresponding aldehydes in acetonitrile solution.31
On the other hand, we found that pure TiO2 exhibits efficient and selective oxidation of benzyl alcohol into benzaldehyde with a selectivity of >99% by O2 in acetonitrile solution under visible light irradiation.32–34 This simple photocatalytic system is an energy-saving and green process which can operate under solar light irradiation using O2 as an oxidant. However, to date, the details of the mechanism behind the visible light induced photocatalytic reaction have not yet been investigated. The reaction mechanism on pure TiO2 has, thus, been extensively studied by a combination of spectroscopic techniques and photo-electrochemical measurements. In particular, we report on the characterization of a charge transfer complex formed by the interaction of benzyl alcohol with the TiO2 surface as the active site and the mechanism behind the photocatalytic reaction on the basis of the reaction kinetics.
Experimental
Materials
All chemicals and solvents used in this study were of analytical grade and were utilized without further purification. Acetonitrile (99.5%), normal benzyl alcohol (99.5%) and α,α-d2 benzyl alcohol (99.5%) were obtained from Wako Pure Chemical Industries, Ltd. The TiO2 (ST-01) purchased from Ishihara Co., Ltd was used as the untreated TiO2 photocatalyst, which involves only the anatase structure. Two different surface modifications of the TiO2 (ST-01) were performed:33 1) TiO2 was heated at a ramp of 5 K min−1, and kept at 673–973 K for 3 h in an electric furnace in air. For example, the sample heat-treated at 673 K is referred to as TiO2(673); 2) The diluted HF solutions (0.006–0.4 wt%) were prepared in water (50 mL) involving different volumes (6–400 μL) of 50 wt% aq. HF. The TiO2 (1.0 g) was suspended in the dil. HF solution for 2 h in a Teflon beaker. After HF treatment, the samples were washed in distilled water, filtrated and dried at 343 K for 12 h. The presence of F− ions in the solution separated from the TiO2 suspension was determined by the addition of sat. CaCl2 solution to form a CaF2 precipitate (limitation detected: >7 ppm). As a consequence, F− ions could be hardly detected in the solution for all the samples prepared, suggesting that they were mostly adsorbed on the TiO2 surface. The HF-treated TiO2 were then referred to as HF(x)–TiO2 (x: 6–400). The x stands for the volume (μL) of 50 wt% aq. HF added to the TiO2 suspension.
Photocatalytic reactions
Photocatalytic reactions were carried out in a pyrex cell (20 mL in volume). The TiO2 photocatalyst (50 mg) was suspended in acetonitrile solution (10 mL) involving benzyl alcohol (50 μmol) in the cell and the gas phase was purged by pure O2 (1 atm). Photo-irradiation was performed after confirming the equilibrium adsorption of benzyl alcohol for 1 h under dark conditions. A blue LED lamp (OptiLED, 2.5 W, IMPACT EYE Co. Ltd.) was used for photo-irradiation, the energy profile of which is shown in a previous report.32 The photo-intensity of the blue LED lamp was measured by an illumination meter (TMS 870, TASCO Co., Ltd.) which was adjusted to 1.8 × 104 lux. After the reaction, the catalysts were immediately separated from the solution by filtration through a 0.20 μm membrane filter (Dismic-25, Advantec). The solution was then analyzed by high performance liquid chromatography: HPLC (Shimadzu LC10ATVP, UV-Vis detector, column: Chemcopak, mobile phase: a mixture of acetonitrile and 1.0 vol.% aqueous formic acid) and the gas phase (CO2) was analyzed by gas chromatography (TCD): GC (Model-802, Ohkura; column: porapak Q). The presence of H2O2 in the reaction liquid was examined by monitoring the UV-Vis absorbance at 407 nm after the addition of titanium(IV) oxysulfate solution in dilute sulfuric acid (Aldrich).
Quantum yields
The photo-oxidation of benzyl alcohol into benzaldehyde was performed on TiO2 (10 mg) in acetonitrile solution (2 mL) involving 10 μmol of benzyl alcohol in a quartz cell using a 500 W xenon lamp (Ushio, Optical Modulex) through a monochromator (Koken, SG-100). The full width at half maximum (FWHM) of each photo-irradiated wavelength was ca. 13.6 nm and the photo-energy was measured by a power-meter (Ophir, ORION/PD). The photon energy was adjusted to a photon flow of 0.5–0.8 mW cm−2, as measured by a power meter (ORION/PD, Ophir). The quantum yield (Φ) is defined in the following eqn (1):| |  | (1) |
Reaction kinetics
The reaction rate constant for the photocatalytic oxidation of benzyl alcohol into benzaldehyde was evaluated from the first-order rate law in eqn (2) and eqn (3):| | 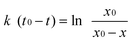 | (2) |
| | | x = x0 {1- exp k (t0-t)} | (3) |
Here, x0, x, k, t and t0 represent the amounts of added benzyl alcohol (50 μmol), photo-formed benzaldehyde, the reaction rate constant, reaction time and induction period, respectively.
The activation energy for the photocatalytic reaction was estimated by the Arrhenius plot from eqn (4).
| | 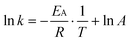 | (4) |
Here, EA, R, T and A represent the activation energy, gas constant, absolute temperature and pre-exponential factor, respectively.
Characterizations
UV-Vis spectroscopic measurements of the samples were carried out by diffused reflectance mode using a UV-Vis spectrophotometer (UV-3100PC, Shimadzu). The atomic composition of the HF-treated TiO2 was analyzed by X-ray photoelectron spectroscopy (XPS, ESCA 3200, Shimadzu). The infrared spectra of the pure TiO2 or TiO2 adsorbed with benzyl alcohol were recorded in absorbance mode with a FT-IR (JASCO FT/IR 620), and measured with 100 scans at 4 cm−1 resolution. Prior to measurements, a self-supporting pellet of the TiO2 photocatalyst was heat-treated under O2 at 673 K for 6 h and then evacuated at 523 K for 30 min in the cell. The adsorption of benzyl alcohol on the heat-treated TiO2 was carried out by introducing the well-evacuated benzyl alcohol vapor (ca. 25 Pa) into the cell at 298 K and the gas phase was then evacuated after adsorption was complete. The spectrum of the adsorbed benzyl alcohol was obtained by subtracting the spectrum of TiO2 from that of the benzyl alcohol-adsorbed TiO2.
Electrochemical measurements
Photo-electrochemical measurements were carried out in a three electrode cell equipped with a quartz window. A TiO2 film, Pt wire and saturated KCl Ag/AgCl electrode were used as working electrode, counter electrode and reference electrode, respectively. The acetonitrile solution involving 5 mM benzyl alcohol and 0.1 M LiClO4 was used as the electrolyte solution. Working electrodes were fabricated as follows: a TiO2 (1.0 g) was well-dispersed in water (2 mL) and the paste was then spread onto an ITO-glass electrode (10 Ω cm−2) by a squige method, followed by heat-treatment at 473 K for 30 min in air. The film mass of electrodes was adjusted to 1.20 ± 0.050 mg cm−2. Photo-irradiation of the TiO2 film was performed under visible light irradiation with the 500 W Xenon lamp (Optical Modulex SX-UI501XQ, Ushio Inc.) through different low cut-off filters (Asahi Technoglass Co. Ltd.). The flatband potential of the TiO2 film was obtained by the Mott-Schottky plots using a potentiostat (HZ3000, Hokuto Denko).
Results
We performed the selective photocatalytic oxidation of benzyl alcohol to benzaldehyde by O2 using TiO2 photocatalyst under visible light irradiation at ambient temperature. The effect of heat-treatment on TiO2 and its chemical modification with hydrofluoric acid were extensively investigated in order to clarify the active sites for the photocatalytic oxidation of benzyl alcohol.
Effect of heat-treatment temperature on TiO2
The TiO2 heat-treated at different temperatures were used as photocatalysts and the TiO2 samples heated at ∼973 K possess anatase structures as shown in Fig. S.1 (ESI†). The reaction time profiles for the selective oxidation of benzyl alcohol under visible light irradiation are shown in Fig. S.2 (a–e).† Before irradiation at t = 0, only an equilibrium adsorption of benzyl alcohol on TiO2 was observed. The amount of benzyl alcohol adsorbed on TiO2 depended on the heat-treatment temperature of TiO2, and it decreased with an increase in the heat-treatment temperature. When photo-irradiation of visible light was started, the amount of benzyl alcohol decreased with an increase in time, while the amount of benzaldehyde increased with high selectivity (>99%) as shown in Fig. S.2 (a).† The reaction rate constant (k) for the formation of benzaldehyde was approximated by the first order equation using eqn (2) and eqn (3). An induction period (t0) was introduced for a better fitting, and it may be attributed to the surface reconstruction between benzyl alcohol and TiO2 during photo-irradiation. The t0 and k for the photocatalytic reaction were determined as shown in Fig. S.2 (a′–e′). Fig. 1 shows the dependence of the heat-treatment temperature on the photocatalytic activities and the adsorbability of benzyl alcohol on the TiO2 photocatalysts. As shown in Fig. 1, it was found that the activities for the formation of benzaldehyde and uptakes of benzyl alcohol in the dark decrease on the TiO2 with an increase in the heat-treatment temperature. These results indicate that benzyl alcohol adsorbed on the TiO2 can be, thus, a vital role in the photocatalytic reaction.
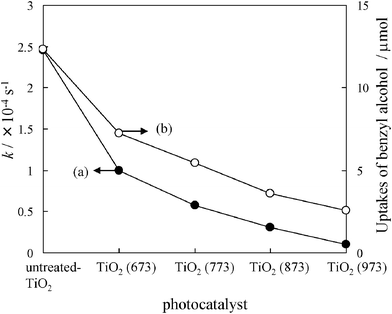 |
| | Fig. 1 Reaction rate constants (k) for the selective photocatalytic oxidation of benzyl alcohol (a) and the uptakes of benzyl alcohol (b) on the heat-treated TiO2 at different temperatures. | |
Effect of HF-treatment on TiO2
The effect of HF treatment of TiO2 on the photocatalytic activity was investigated. Fig. 2 shows the photocatalytic activities for the oxidation of benzyl alcohol as a function of the amount of HF on the different heat-treated TiO2. As shown in Fig. 2 (a), the photocatalytic activity decreased with an increase of amounts of HF added to the TiO2 sample, and its activity was hardly observed on the TiO2 treated by 62.5 μL of aq. HF. These results indicate that the interaction of F− ions with TiO2 causes a decrease in the active sites for the photocatalytic reactions. Likewise, TiO2(773) and TiO2(973) modified with HF exhibited a decrease in the photocatalytic activity, as shown in Fig. 2 (b, c), while the TiO2(773) and TiO2(973) were markedly deactivated with less HF added than the untreated TiO2. According to our previous report,33 the BET surface area of TiO2 was estimated to be 320 m2 g−1 for untreated TiO2, 158 m2 g−1 for TiO2(773), and 91 m2 g−1 for TiO2(973). From these results, it was found that the number of surface active sites decrease as the BET surface area of TiO2 decreases.
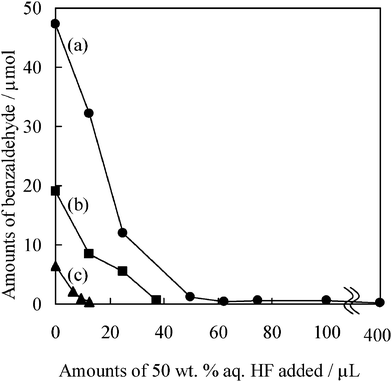 |
| | Fig. 2 Photocatalytic activities for the formation of benzaldehyde on various TiO2 samples under visible light irradiation for 4 h. Surface treatment by 50 wt% aq. HF was conducted on: (a) untreated TiO2, (b) TiO2(773) and (c) TiO2(973). | |
Surface characterizations
In order to clarify the origins of the visible light induced photocatalysis, characterization studies were performed by UV-Vis, FT-IR and XPS spectroscopies. Fig. 3 (a, b) shows the UV-Vis spectra of the pure TiO2 and the TiO2 adsorbed with benzyl alcohol, respectively.35 It was observed that the TiO2 adsorbed with benzyl alcohol exhibits visible light absorption, while the TiO2 exhibits little visible light absorbance. The visible light absorbance is not due to a direct band-gap excitation (3.1 eV) but to the excitation of the CT complex formed by the interaction of TiO2 with benzyl alcohol. Fig. 3 (c) shows the quantum yields for the formation of benzaldehyde in the photocatalytic oxidation of benzyl alcohol on TiO2 using eqn (1). It was observed that the photocatalytic activities are extended towards ca. 700 nm, and the action spectra correspond with the absorbance of the surface complex. These results indicate that the photo-excitation of the surface complex by itself plays a significant role in the photo-oxidation of benzyl alcohol.
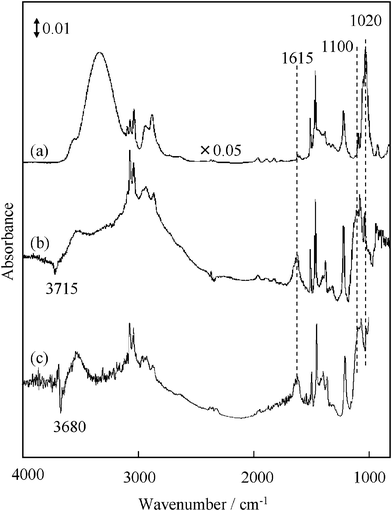
Fig. 4 shows FT-IR spectra of the untreated TiO2, TiO2 heat-treated at different temperatures and TiO2 modified with HF. As shown in Fig. 4 (a), the untreated TiO2 exhibits the un-resolved IR band at ca. 3500–3700 cm−1, which are due to the surface OH groups involving weakly adsorbed water molecules. When the TiO2 was heat-treated at 673 K in O2, the IR bands at 3636, 3680 and 3715 cm−1 were clearly observed (Fig. 4 (b)). Upon calcination of TiO2 at 773 K, the IR band at 3636 cm−1 disappeared and the intensity of the bands at 3680 and 3715 cm−1 decreased (Fig. 4 (c)). Furthermore, the IR band at 3680 cm−1 almost disappeared at 873 K, while that at 3715 cm−1 is still observed (Fig. 4 (d)). According to a previous report,36–38 the IR band at 3636 cm−1 can be assigned to the physically adsorbed H2O on the surface, while the other type of OH groups were identified as follows: the band at 3680 cm−1 corresponds to the adjacent OH groups interacting via weak hydrogen bonds (bridge OH groups bound to two other Ti sites), while the band at 3715 cm−1 can be attributed to the terminal OH groups. From the XPS analysis,33 the surface state of HF-treated TiO2 was confirmed that the F− ions are adsorbed on the TiO2 surface and the surface OH groups of TiO2 is partially modified by HF possibly to form ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti–F or TiOF2.39 As shown in Fig. 4 (e), the IR band at 3715 cm−1 on the HF(50) –TiO2 drastically decreased, while the significant change of the IR band at 3680 cm−1 was not observed, compared with the TiO2 untreated by HF. Furthermore, HF(400)–TiO2 also showed a band at 3680 cm−1 and its peak intensity was lower than that of HF(50)–TiO2, as shown in Fig. 4 (f). These results indicate that the terminal Ti–OH groups sensitively react with the appropriate amount of HF, while the bridge OH groups react with an excess of HF.
Ti–F or TiOF2.39 As shown in Fig. 4 (e), the IR band at 3715 cm−1 on the HF(50) –TiO2 drastically decreased, while the significant change of the IR band at 3680 cm−1 was not observed, compared with the TiO2 untreated by HF. Furthermore, HF(400)–TiO2 also showed a band at 3680 cm−1 and its peak intensity was lower than that of HF(50)–TiO2, as shown in Fig. 4 (f). These results indicate that the terminal Ti–OH groups sensitively react with the appropriate amount of HF, while the bridge OH groups react with an excess of HF.
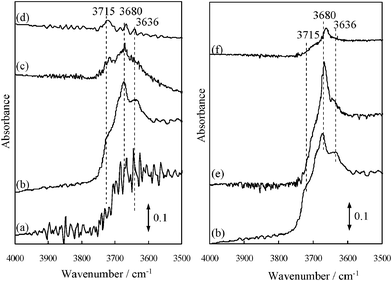 |
| | Fig. 4
FT-IR spectra of the untreated TiO2 (a), TiO2 heat-treated at: (b) 673 K, (c) 773 K, (d) 873 K; (e) HF(50)–TiO2 and (f) HF(400)–TiO2. The spectra (e) and (f) were recorded after heat-treatment at 673 K. | |
Moreover, the states of benzyl alcohol adsorbed on TiO2 were analyzed by FT-IR spectroscopy and the results are shown in Fig. 5. It was observed that the IR band of the benzyl alcohol adsorbed on TiO2 is essentially the same as that for the molecular benzyl alcohol by itself (Fig. 5 (a, b)). However, the following unique features caused by the interaction of benzyl alcohol with the TiO2 surface are observed in Fig. 5 (b): (i) a remarkable downward negative band at 3715 cm−1 attributed to the O–H stretching of the terminal OH group; (ii) a marked increase in the band intensity at 1615 cm−1 attributed to the phenyl ring vibration; and (iii) a new band appears at ca. 1100 cm−1, which is attributed to the C–O stretching of the alcolate species formed between TiO2 and benzyl alcohol, while that of benzyl alcohol by itself is 1020 cm−1. It should be noted that the similar alcolate species were previously reported in a system of Nb2O5 adsorbed with alcohol.40 On the other hand, HF(400)–TiO2 adsorbed with benzyl alcohol also showed a C–O stretching mode (1100 cm−1) due to the alcolate, while a negative band was observed at 3680 cm−1 instead of one at 3715 cm−1, as shown in Fig. 5 (c). Therefore, it was confirmed that the alcolate is formed on the untreated TiO2 as well as on the HF-treated TiO2. Taking the results of Fig. 2 into consideration, the surface alcolate species formed by the interaction of benzyl alcohol with the terminal OH groups, clearly plays a significant role in the photocatalytic reaction while the interaction with the bridge OH groups are inactive for the reaction.
Reaction kinetics
The dependence of the reaction temperature on the photocatalytic activities was investigated and the results are shown in Fig. 6. When the photocatalytic oxidation of benzyl alcohol or α,α-d2 benzyl alcohol was carried out on TiO2 at different temperatures (283–313 K), it was observed that the activities for the formation of corresponding benzaldehyde increased with an increase of the reaction temperature as shown in Fig. 6 [I] and [II], respectively. It should be noted that the reaction little proceeds at 283–313 K in the dark. The amount of photo-formed benzaldehyde was simulated from the eqn (2) and (3), and the reaction rate constant (k) and induction period (t0) are also shown in Fig. S.3 (ESI).† From the simulations, the kinetic isotope effect (KIE) [= kC–H/kC–D] was estimated to be ≈4.0 at 283 K. Therefore, it was confirmed that the process for the α-deprotonation is the rate determining step (RDS) for the overall reaction. Moreover, it was observed that the KIE decreases from 4.0 to 2.8 as the reaction temperature increases from 283 to 313 K. The Arrhenius plots are shown in Fig. 6 [III], and the activation energy (EA) was determined to be 19 kJ mol−1 for normal benzyl alcohol and 28 kJ mol−1 for α,α-d2 benzyl alcohol using eqn (4).
Discussions
It was confirmed that the terminal OH groups on the TiO2 surface play a significant role in the photocatalytic reactions by FT-IR analysis. From the results of Fig. 2, the number of terminal OH groups was roughly estimated.41 As shown in Fig. 7, the untreated TiO2 showed the highest reaction rate among various photocatalysts, and it was quantitatively determined that the photocatalytic activities decrease as the number of terminal OH groups on the TiO2 decrease. Moreover, the density of terminal OH groups on the untreated TiO2 was calculated to be ca. 3.4 nm−2, which is in a good agreement with a previous report.37 When the photo-oxidation of benzyl alcohol (10 mmol) on TiO2 (50 mg) was performed under visible light irradiation, 2.2 mmol of benzaldehyde was produced after 36 h.32 Considering the density of terminal OH groups (3.4 nm−2), the turnover number (TON) and turnover frequency (TOF) was estimated to be ca. 24 and 0.7 h−1, respectively, suggesting that this reaction does, indeed, take place photocatalytically.
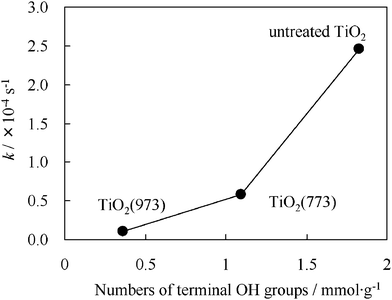 |
| | Fig. 7 Relationship between the reaction rate constants (k) and the number of terminal OH groups on various heat-treated TiO2. | |
In order to understand the initial electron transfer on the CT complex, the photo-reducibility of the Ag+ ions as an electron acceptor was investigated on TiO2 under visible light irradiation in the absence of O2.42UV-Vis spectra of TiO2 adsorbed with Ag+ ions in the presence of normal benzyl alcohol and α,α-d2 benzyl alcohol are shown in Fig. 8 [I and II], respectively. It was observed that the absorbance in the region of 400–800 nm increases with an increase in the irradiation time. This absorbance is attributed to the formation of Ag particles by the reduction of Ag+ ions.43 It should be noted that the photo-reduction of Ag+ ions takes place neither in the absence of benzyl alcohol nor TiO2 under visible light irradiation. The reaction rate constant (k′) for the photo-reducibility of Ag+ ions was evaluated from the first order equation.44 The photo-reduction rate constants of Ag+ ions in the presence of normal benzyl alcohol and α,α-d2 benzyl alcohol were determined to be k′H = 4.87 × 10−3 s−1 and k′D = 1.11 × 10−3 s−1 for (a) and (b), respectively (cf.Fig. 8 [IV]). That is, the KIE for the photo-reducibility of Ag+ ions (k′H/D) was determined to be 4.4, closely associated with the value (kH/D = 3.9) for the photocatalytic oxidation of benzyl alcohol into benzaldehyde at 298 K.32 Therefore, it can be seen that the efficiency for the electron transfer to the electron acceptors such as Ag+ ions and O2 molecules correlates with the α-deprotonation ability from benzyl alcohol on the CT complex. On the other hand, HF-treated TiO2 did not exhibit any significant change in the absorbance (Fig. 8 [III]). That is, the modification of terminal OH groups on the TiO2 with HF cannot induce the efficient electron transfer to the electron acceptors.
![Changes in the UV-Vis spectra of untreated TiO2 adsorbed with: [I] normal benzyl alcohol, [II] α,α-d2 benzyl alcohol, and [III] HF(50)-TiO2 adsorbed with normal benzyl alcohol in the presence of Ag+ ions under visible light irradiation, and the plots [IV] for ln [A0/(A0 − A)] against t for: (a) normal benzyl alcohol, and (b) α,α-d2 benzyl alcohol.](/image/article/2012/RA/c1ra00417d/c1ra00417d-f8.gif) |
| | Fig. 8 Changes in the UV-Vis spectra of untreated TiO2 adsorbed with: [I] normal benzyl alcohol, [II] α,α-d2 benzyl alcohol, and [III] HF(50)-TiO2 adsorbed with normal benzyl alcohol in the presence of Ag+ ions under visible light irradiation, and the plots [IV] for ln [A0/(A0 − A)] against t for: (a) normal benzyl alcohol, and (b) α,α-d2 benzyl alcohol. | |
The EA was determined to be 19 kJ mol−1 for normal benzyl alcohol and 28 kJ mol−1 for α,α-d2 benzyl alcohol under visible light irradiation (E < 2.7 eV), suggesting that the isotope effect offers an influence to the activation energy. According to the previous report, the EA was determined to be 21 ± 1 kJ mol−1 in the photocatalytic oxidation of benzyl alcohol by O2 on TiO2 under UV-light irradiation (E > 3.2 eV).45 The EA, therefore, does not significantly depend on the photo-energy. It was observed that neither the adsorbability of benzyl alcohol nor benzaldehyde significantly changes at various temperature (283–313 K) on the TiO2. Taking these results into consideration, the EA is, indeed, associated with the α-deprotonation assisted by the terminal OH group of TiO2 after photo-excitation.
In order to understand the energy diagram in the photocatalytic system, photo-electrochemical measurements were performed. As shown in Fig. 9 [I], a stable anodic photocurrent was observed on the TiO2 film at E = + 0.6 V vs.Ag/AgCl (pH = 3.8) under λ > 420 nm although the photocurrent efficiency is low. The Jph–V characteristics on the TiO2 indicate that the photocurrent efficiency in the presence of benzyl alcohol is higher than in its absence as shown in Fig. 9 [II]. Therefore, the anodic photocurrent was observed to participate in the oxidation of benzyl alcohol, while the onset potential was estimated to be ca. + 0.25 V vs.Ag/AgCl. Fig. 9 [III] shows the effect of the photo-irradiated light wavelength on the photocurrent in the presence of benzyl alcohol. It was observed that the photo-response was extended towards 600 nm, and the photocurrent increases as the light wavelength shortens. On the other hand, the Mott-Schottky plots (1/C2vs. E) for the TiO2 film electrode in the presence of benzyl alcohol showed the flatband potential at ca. −0.80 V vs.Ag/AgCl (pH = 3.8), as shown in Fig. S.4 (ESI).† It was previously reported that the oxygen vacancies are located ca. 1.0 V below the conduction band of TiO2.46 Therefore, even if the electrons are excited into the conduction band of the TiO2 under visible light irradiation, the photo-induced electrons are mostly trapped on the oxygen vacancies where molecular O2 would be readily reduced when considering a redox potential of O2/OH−: E = +0.8 V vs.Ag/AgCl (pH = 3.8).47 When the TiO2 adsorbed with benzyl alcohol in the absence of O2 was illuminated by visible light,48 the absorbance at 400–800 nm increased as shown in Fig. 10 (a, b). This absorbance can be attributed to the electron trappings on the oxygen vacancies (color center).49 Subsequently, exposure of O2 into the photo-reduced sample leads to a drastic decrease of the absorbance, as shown in Fig. 10 (c), which is due to the electron transfer from the color center to O2 to form superoxide. The photo-formed benzyl alcoholic radical would be further deprotonated by superoxide, which is known to be a base that accepts protons.50
![Photocurrent [I] on the TiO2 film in the presence of benzyl alcohol under λ > 420 nm, Jph–V characteristics [II] in the presence (a) and absence (b) of benzyl alcohol under λ > 420 nm; and dependence [III] of the light wavelength photo-irradiated on the photocurrent. The spectra in [I] and [III] were recorded at +0.6 V vs.Ag/AgCl.](/image/article/2012/RA/c1ra00417d/c1ra00417d-f9.gif) |
| | Fig. 9 Photocurrent [I] on the TiO2 film in the presence of benzyl alcohol under λ > 420 nm, Jph–V characteristics [II] in the presence (a) and absence (b) of benzyl alcohol under λ > 420 nm; and dependence [III] of the light wavelength photo-irradiated on the photocurrent. The spectra in [I] and [III] were recorded at +0.6 V vs.Ag/AgCl. | |
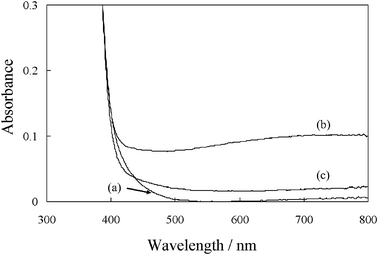 |
| | Fig. 10 Change in the absorption spectra of untreated TiO2 adsorbed with benzyl alcohol under: (a) dark conditions, (b) visible light irradiation for 2 h and (c) after subsequent exposure of O2 into (b). | |
It should be noted that H2O2 molecules were not detected in the reaction liquid during the photocatalytic reaction, while the photooxidation of benzyl alcohol into benzaldehyde scarcely proceeded on the TiO2 sample in the presence of H2O2 under visible light irradiation.51 According to a previous report,16 treatment with Brønsted acids can lead to significant rate enhancement for the oxidation of alcohols on TiO2 under UV-light irradiation due to the acceleration of a disproportionate decomposition of the side-on peroxide species on the TiO2. However, our photocatalytic system demonstrates that the presence of the Brønsted acids (H2SO4) did not significantly enhance the photocatalytic activity under visible light irradiation, as shown in Fig. S.5 (ESI),† supporting the direct regeneration of the surface OH group by the reduction of O2, not being by way of the formation of H2O2 as an intermediate.
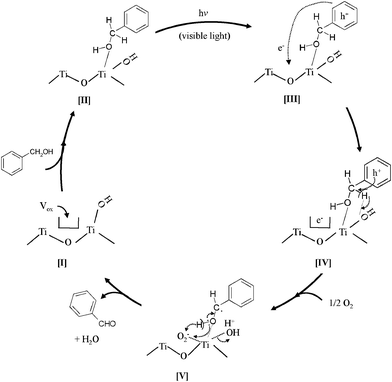
Taking these results into consideration, the reaction mechanism behind the selective photocatalytic oxidation of benzyl alcohol into benzaldehyde by O2 under visible light irradiation is depicted in Scheme 1. The TiO2 involves intrinsic surface oxygen vacancies (Vox) closely associated with the terminal OH group in order to compensate the charge balance (Scheme 1 [I]).37 When the benzyl alcohol is adsorbed on TiO2, the alcolate species (CT complex) is formed by the interaction of the α-OH group of benzyl alcohol with a Ti site (Scheme 1 [II]). This interaction induces absorption in the visible light region which was assigned to the ligand-to-metal charge transfer (LMCT) of the surface CT complex as shown in Scheme 1 [III], based on the previous reports52,53 visible light irradiation of the surface CT complex produces hole and electron, and the hole induces the α-deprotonation from the benzyl alcohol by an assist of the terminal OH as shown in Scheme 1 [IV]. Subsequently, the water molecule is released from the TiO2 surface, while the electron is transferred to O2 as shown in Scheme 1 [V]. Such superoxide species would further induce the deprotonation from the CT complex to form benzaldehyde, accompanied by the regeneration of the surface terminal OH groups. The consecutive generation of the terminal OH groups would, thus, be one of the key-factors for the photocatalytic reaction.
Conclusions
The oxidation of benzyl alcohol by O2 was investigated on the TiO2 photocatalyst under visible light irradiation at room temperature. It was found that benzyl alcohol is converted into benzaldehyde with high selectivity (>99%). The origin of the visible light-induced photocatalysis was investigated using UV-Vis, FT-IR and XPS spectroscopies. It was found that the charge transfer surface complex, which is formed by the interaction of benzyl alcohol with TiO2, is excited under visible light irradiation. The rate determining step (RDS) in this reaction is the α-deprotonation from benzyl alcohol, the process of which is assisted by the terminal OH groups of the TiO2. The activation energy was determined to be 19 kJ mol−1. The reaction mechanism behind the selective photocatalytic oxidation of benzyl alcohol into benzaldehyde on TiO2 was clearly demonstrated by a combination of photoelectrochemical investigations. These findings further contribute to an understanding of surface CT complex mediated photocatalysis, and to providing a sustainable pathway for green organic synthesis under solar light energy.
References
- A. Fujishima, T. N. Rao, D. A. Tryk and J. Photochem, Photobiol. C: Photochem. Rev., 2000, 1, 1 Search PubMed.
- S. Sato and J. M. White, J. Catal., 1981, 69, 128 CrossRef CAS.
- M. Grätzel and J. Photochem, Photobiol. C: Photochem. Rev., 2003, 4, 145 Search PubMed.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- Y. Shiraishi, T. Hirai and J. Photochem, Photobiol., C: Photochem. Rev., 2008, 9, 157 Search PubMed.
- G. Palmisano, E. García-López, G. Marcì, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46, 7074 RSC.
- Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656 CAS.
- G. Zhang, J. Yi, J. Shim, J. Lee and W. Choi, Appl. Catal., B: Environ., 2011, 102, 132 Search PubMed.
- J. T. Carneiro, C. C. Yang, J. A. Moulijn and G. Mul, J. Catal., 2011, 277, 129 Search PubMed.
- J. T. Carneiro, T. J. Savenije, J. A. Moulijn, G. Mul and J. Photochem, Photobiol., A: Chem., 2011, 217, 326 Search PubMed.
- J. T. Carneiro, J. A. Moulijn and G. Mul, J. Catal., 2010, 273, 199 CrossRef CAS.
- A. R. Almeida, J. T. Carneiro, J. A. Moulijn and G. Mul, J. Catal., 2010, 273, 116 CrossRef CAS.
- J. T. Carneiro, A. R. Almeida, J. A. Moulijn and G. Mul, Phys. Chem. Chem. Phys., 2010, 12, 2744 RSC.
- S. J. Song, K. S. Kim, K. H. Kim, J. B. Kim, J.H. Kim, K.S. Kim, H. Shin and D.L. Cho, Chem. Lett., 2008, 37, 1052 Search PubMed.
- H. Kominami, H. Sugahara and K. Hashimoto, Catal. Commun., 2010, 11, 426 CrossRef CAS.
- Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976 CrossRef CAS.
- M. Zhang, Q. Wang, C. Chen, L. Zang, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081 CrossRef CAS.
- O. S. Mohamed, A. E. M. Gaber, A. A. Abdel-Wahab and J. Photochem, Photobiol. A, 2002, 148, 205 Search PubMed.
- S. Farhadi, M. Afshari, M. Maleki and Z. Badazadeh, Tetrahedron Lett., 2005, 46, 8483 CrossRef CAS.
- A. Z. G. G. Fattima, N. A. Ahmed and H. H. Falah, Desalination, 2009, 209, 342 Search PubMed.
- U. R. Pillai and E. Sahle-Demessie, J. Catal., 2002, 211, 434 CrossRef CAS.
- V. Augugliaro and L. Palmisano, ChemSusChem., 2010, 3, 1135 Search PubMed.
- S. Yurdakal, G. Palmisano, V. Loddo, O. Alagoez, V. Augugliaro and L. Palmisano, Green Chemistry, 2009, 11, 510 RSC.
- V. Augugliaro, V. Loddo, M. J. Lopez-Munoz, C. Marquez-Alvarez, G. Palmisano, L. Palmisano and S. Yurdakal, Photochem. Photobiol.
Sci., 2009, 8, 663 RSC.
- S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568 CrossRef CAS.
- V. Augugliaro, T. Caronna, V. Loddo, G. Marci, G. Palmisano, L. Palmisano and S. Yurdakal, Chem. Eur. J., 2008, 14, 4640 CrossRef CAS.
- G. Palmisano, S. Yurdakal, V. Augugliaro, V. Loddo and L. Palmisano, Adv. Synth. Catal., 2007, 349, 964 CrossRef CAS.
- G. Palmisano, M. Addamo, V. Augugliaro, T. Caronna, E. Garcia-Lopez, V. Loddo and L. Palmisano, Chem. Commun., 2006, 1012 RSC.
- M. Addamo, V. Augugliaro, M. Bellardita, A. D. Paola, V. Loddo, G. Palmisano, L. Palmisano and S. Yurdakal, Catal. Lett., 2008, 126, 58 CrossRef CAS.
- S. Naya, A. Inoue and H. Tada, J. Am. Chem. Soc., 2010, 132, 6292 CrossRef CAS.
- L. Samiolo, M. Valigi, D. Gazzoli and R. Amadelli, Electrochim. Acta, 2010, 55, 7788 Search PubMed.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279 CrossRef CAS.
- S. Higashimoto, K. Okada, T. Morisugi, M. Azuma, H. Ohue, T. H. Kim, M. Matsuoka and M. Anpo, Top. Catal., 2010, 53, 578 CrossRef CAS.
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76 CrossRef CAS.
- The sample for the UV-Vis analysis was prepared as follows: The TiO2 powder (0.5 g) was suspended in acetonitrile solution (100 mL) involving 5.0 mM benzyl alcohol for 2 h under dark conditions, followed by separation and drying in air at 298 K.
- A. Y. Nosaka, J. Nishino, T. Fujiwara, T. Ikegami, H. Yagi, H. Akutsu and Y. Nosaka, J. Phys. Chem. B, 2006, 110, 8380 Search PubMed.
- G. D. Parfitt, Prog. Surf. Membr. Sci., 1976, 11, 181 Search PubMed.
- K. V. R. Chary, T. Bhaskar, G. Kishan and V. Vijayakumar, J. Phys. Chem. B, 1998, 102, 3936 CrossRef CAS.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. Lu and H. M. Cheng, Chem. Commun., 2010, 46, 755 RSC.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi, H. Yamashita and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713 CrossRef CAS.
- The number of terminal OH groups were estimated from the amounts of F− ions, which were consumed for the deactivation of the photocatalyst. For example, the untreated TiO2 (1.0 g) requires 62.5 μL of 50 wt% aq. HF for the catalyst deactivation (see Fig. 2), i.e., the number of terminal OH groups were estimated to be ca. 1.8 mmol/1 g-TiO2. Moreover, from the surface area (320 m2 g−1), the density of the terminal OH groups were calculated to be ca. 3.4 nm−2.
- The sample for the UV-Vis analysis was prepared as follows: The TiO2 (100 mg) was spread on the sample folder (20 × 15 × 0.5 mm). The sample was impregnated with 0.2 mL of acetonitrile involving 0.25 M benzyl alcohol (50 μmol) and 0.25 M AgNO3 (50 μmol), and was then sealed with quartz in N2 purged globebox.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928 CrossRef CAS.
- The photo-reducibility of Ag+ ions on TiO2 was evaluated by the following first-order equation: k′t = ln{A0/(A0 − A)}. Here, k′, A and A0 represent the rate constant for photo-reducibility, absorbance at 500 nm as a function of time, and with the saturated spectrum after prolonged irradiation, respectively. The A and A0 were calculated by subtracting the spectrum after visible light irradiation with
that before illumination.
- F. A. Z. G. Gassim, A. N. Alkhateeb and F. H. Hussein, Desalination, 2007, 342 Search PubMed.
- S. Higashimoto and M. Azuma, Appl. Catal. B: Env., 2009, 89, 557 Search PubMed.
-
A. Sobkowiak and J. L. Roberts, in Electrochemistry for Chemists, ed. D. T. Sawyer, John Wiley & Sons, Inc., New York, 1996 Search PubMed, translated in Japanese.
- The sample for the UV-Vis analysis was prepared as follows: The TiO2 (100 mg) was spread on the sample folder (20 × 15 × 0.5 mm). The sample was impregnated with 0.2 mL of acetonitrile involving 0.25 M benzyl alcohol, and was then sealed with quartz in N2 purged glovebox.
- V. N. Kuznetsov and N. Serpone, J. Phys. Chem. C, 2009, 113, 15110 CrossRef CAS.
-
Organic Electrochemistry, ed. H. Lund and O. Hammerich, Marcel Dekker, New York, 2001 Search PubMed.
- Photooxidation of benzyl alcohol (50 μmol) was performed in the presence of H2O2 (100 μmol) on TiO2 (50 mg) under visible light irradiation for 4 h.
- Y. Wang, K. Hang, N. A. Anderson and T. Lian, J. Phys. Chem. B, 2003, 107, 9434 CrossRef CAS.
- T. Tachikawa, S. Tojo, M. Fujitsuka and T. Majima, Langmuir, 2004, 20, 2753 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the reaction time profiles, effect of Brønsted acid on the reaction, and the Mott-Schottky plots for TiO2 in the presence of benzyl alcohol. See DOI: 10.1039/c1ra00417d |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 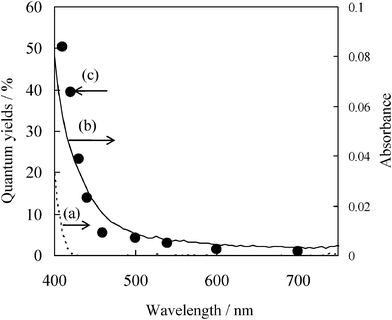
![Time profile for the photocatalytic oxidation of [I] normal benzyl alcohol, [II] α,α-d2 benzyl alcohol on TiO2 at 283–313 K under visible light irradiation, and the Arrhenius plots [III] for (a) benzyl alcohol and (b) α,α-d2 benzyl alcohol.](/image/article/2012/RA/c1ra00417d/c1ra00417d-f6.gif)