Biosurfactant-mediated one-step synthesis of hydrophobic functional imogolite nanotubes
Bejoy
Thomas
abc,
Thibaud
Coradin
abc,
Guillaume
Laurent
abc,
Romain
Valentin
d,
Zephirin
Mouloungui
d,
Florence
Babonneau
abc and
Niki
Baccile
*a
aUPMC Univ Paris 06, UMR 7574, Chimie de la Matière Condensée de Paris, F-75005, Paris, France. E-mail: niki.baccile@upmc.fr; Fax: (+33) 144271544
bCNRS, UMR 7574, Chimie de la Matière Condensée de Paris, F-75005, Paris, France
cCollège de France, UMR 7574, Chimie de la Matière Condensée de Paris, F-75005, Paris, France
dUnité de Chimie Agro-Industrielle, UMR 1010 INRA/INP, ENSIACET, 4 allée Emile Monso, F-31432, Toulouse, France
First published on 4th November 2011
Abstract
Imogolites are aluminosilicate nanotubes that attract a particular interest due to their insulating properties, surface reactivity and shape-selective properties in catalysis. Although being available in nature, synthetic protocols have been developed since the 70's to obtain large amounts of this particular mineral without purification processes. In particular, surface functionalized imogolites are currently studied for their application as a dispersant in hydrophobic matrixes like polymers. With respect to known synthetic processes, natural imogolite is produced by using either milli- or deci-molar amounts of aluminium salts in solution while surface functionalized imogolites are systematically prepared in two steps: imogolite synthesis followed by surface functionalization mainly using phosphonic acids. Here, we propose a new one-step synthesis involving a cheap, widely-used, non-toxic surfactant, glycerol α-monolaurate (MG) and centimolar aluminium concentrations. In the presence of Al3+ cations, MG forms an emulsion, and the subsequent addition of tetraethoxysilane leads to the formation of imogolite nanotubes within 3 days at neutral pH under hydrothermal conditions. An interesting, new, lamellar long-range order of the nanotubes can then be achieved after 10 days. Under similar conditions, blank tests performed on MG-free systems result in the formation of proto-imogolite. Based on the combination of highly selective 29Si, 27Al and 1H solid-state NMR experiments and additional synthesis using lauric acid (LA) and glycerol, a mechanism of formation is proposed based on MG dissociation and LA binding on the outside surface of the nanotubes. As a result, our process provides imogolite with unprecedented hydrophobic surface properties that offer new perspectives for these materials as nanoscale fillers in, e.g., polymeric media.
Introduction
Synthetic metal oxide single-walled nanotubes (SWNTs) are expected to possess a number of interesting and unique properties (surface reactivity, mechanical strength, tailored length, insulating properties, etc.) and have generated a great deal of interest and potential use in a variety of applications such as molecular separation and storage, chemical sensing, drug delivery applications, photonics, biotechnology, catalysis, composite materials and anion/cation retention from water.1 Although much less popular than the conventional carbon nanotubes, they represent attractive materials due to their potentially wide range of tunable compositions and properties accessible by low-temperature liquid-phase chemistry. An important goal in this area is to achieve a precise size control at the nanoscale over the three dimensions to get access to unique properties such as tunable band gaps, ballistic transport of charge/heat/mass, and quantum confinement phenomena.2Although the variety of such SWNTs is not yet extensive, an attractive aspect of these materials is that they can be synthesized by liquid-phase synthesis under moderate conditions, often hydrothermal or solvothermal. An interesting member of this family is imogolite,3 a natural aluminosilicate-based SWNT, whose synthesis has attracted substantial interest in recent years.4,5 The cylindrical wall of the imogolite nanotubes can be visualized as a rolled-up sheet of gibbsite (aluminum hydroxide), with isolated silanol (Si–OH) groups linked to the inner surface of the nanotube wall. The nanotube wall is structurally ordered and is composed of an aluminum octahedron (AlO6) with three oxygen atoms shared by silicon (SiO4) tetrahedra, while the other three oxygen atoms are shared with other Al octahedra in the nanotube wall. The nanotubes have an inner diameter of about 1.0 nm, while the outer diameter of the nanotubes varies with relative composition of Si atoms in the structure. The length of the tubes is in the 400 to 1000 nm range. The imogolite empirical formula was found to be (OH)3Al2O3–SiOH, with [Al]/[Si] = 2. These para-crystalline hydrous silicate polymers are most commonly associated with the weathering of non-crystalline volcanic materials, which have also been identified in pumice deposits, coating of primary particles, stream sediments, and soil derived from igneous and sedimentary rock sandstone. Taken together,3,5 previous reports dedicated to the synthesis, structure and properties of imogolite lead to the following statements: i) synthetic imogolite always appears as SWNTs with a monodispersed diameter size; ii) imogolite formation is generally preceded and/or accompanied by that of proto-imogolite, whose XRD and NMR signatures are indistinguishable from imogolite but whose morphology is not well-defined (particles, platelets); TEM and FT-IR are commonly used to make the distinction between imogolite and proto-imogolite; iii) boehmite and amorphous aluminosilicates are common side-products of the synthetic procedures; iv) even if the theoretical Al/Si molar or atomic ratio is equal to 2, the experimental ratio may vary within a 15% range, depending on the synthesis method; v) nanotube packing occurs in a monoclinic space group rather than in a hexagonal one, as one would expect; vi) the imogolite outer surface is hydrophilic and its isoelectric point is close to pH = 9.
The preparation of the synthetic imogolite generally requires rigorous experimental conditions.4,5 The sol–gel route employing an aluminium salt (mostly aluminium perchlorate or aluminium chloride) in presence of tetraethoxysilane (TEOS) in millimolar concentrations in water at T < 100 °C is the most explored pathway. TEOS is preferred to sodium silicate for its slower condensation kinetics while perchlorates are preferred to chlorides due to their lower complexing ability, both reducing the uncontrolled homo-condensation towards amorphous aluminium hydroxides and silica. The crystallization procedure is optimized at temperatures slightly below 100 °C. Below 90 °C, the formation rate strongly decreases while above 100 °C boehmite rods or gibbsite platelets are formed. The synthesis of pure imogolite from a more concentrated solution is also possible but with usually longer reactions times, going from 10 to 60 days to obtain long-range ordered nanotube packing.5c,d The functionalization of the outer surface of imogolites constitutes a new field of research due to the interest of dispersing imogolite nanotubes in hydrophobic media. Attempts to use organosilanes is not as efficient as expected and phosphonic acids were used instead. Using this procedure, poly(methylmethacrylate) and octadecyl groups were recently grafted on the surface of freshly synthesized imogolite nanotubes providing hydrophobic properties.5h,i So far, no one-pot synthesis of surface modified imogolite has been reported, to the best of our knowledge.
In the present work, we have investigated, for the first time to our knowledge, an alternative hybrid organic/inorganic strategy to control the formation and organization of imogolite SWNTs. Indeed, the use of organic-templating strategies has already been explored to obtain metal oxide nanotubes but all reported structures are multiwalled and have outer diameters which are several tens of nanometres.6 Our organic additive, glycerol α-monolaurate (MG) was selected for two main reasons: 1) monoacylglycerol derivatives (fatty acid chain covalently bonded to a glycerol head group through an ester linkage) are cheap, non toxic, commercial emulsifiers derived from both natural (animal, vegetable) and synthetic routes and routinely used in food products (bakery products, beverages, ice cream, chewing gum, shortening, whipped toppings, margarine, and confections);7–8 2) both glycerol and carboxylic acids can complex Al3+ cations in solution and in the solid state,9 potentially influencing morphological changes and/or process kinetics. In line with our interest to establish a low-environmental impact synthesis, we kept conditions as simple as possible: i) the use of chlorides instead of more toxic perchlorates; ii) the synthesis at quasi-neutral pH and limited pH changes; iii) a centimolar concentration range ([Al3+] > 0.05 mol L−1). The use of TEOS as a silica precursor and the use of hydrothermal (T = 98 °C) conditions are the only factors making our approach similar to previous works.
Through this approach, it is possible to obtain functionalized imogolite SWNTs in less than two weeks under neutral pH conditions. At the moment, boehmite constitutes the main impurity but preliminary results show that an optimization of the synthesis should drastically reduce its overall content. In addition, we show that this process yields surface-modified imogolite nanotubes with hydrophobic properties that make them promising candidates as nanoscale anisotropic fillers in polymer-based composite materials. Finally, surface modification induced an interesting two-dimensional packing of the nanotubes never reported before, as shown by small angle X-ray diffraction and TEM.
Experimental section
Preparation of the materials
Glycerol α-monolaurate (MG) was synthesized following a previously-reported procedure10 on a hydrophobic bed in a continuous reactor at 80 °C giving a high (86%) purity. The resulting powder was equilibrated in 100 mL of distilled water under stirring at room temperature overnight. The desired amount of AlCl3·6H2O was then added under stirring at pH ca. 2.5 at 80 °C. The solution turned into an emulsion and stirring was maintained for 6 additional hours to homogenize the system. Finally, tetraethylorthosilicate (TEOS) was added under stirring and the pH of the system was adjusted to ca. 7 using 1 M NaOH solution about one hour after the TEOS addition. The mixture was allowed to stir at the same temperature for 3 h after which the sample underwent a hydrothermal treatment at 98 °C for a period of 72 h or 240 h. A white solid product was recovered by filtration, washed with distilled water and dried at room temperature under atmospheric conditions. Similar experiments were performed with lauric acid (LA) and glycerol, both purchased from Aldrich. Molar ratios are: additive![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:AlCl3:TEOS = 1:5550:x:3.5 where the additive is MG, LA or glycerol and 1 < x < 7.5. Blank experiments refer to those conducted without any additive.
:H2O:AlCl3:TEOS = 1:5550:x:3.5 where the additive is MG, LA or glycerol and 1 < x < 7.5. Blank experiments refer to those conducted without any additive.
Characterization of the materials
Chemical analysis of the alumina-silica samples was obtained by energy dispersive X-ray spectroscopy (EDAX) using an Oxford X-Max (area: 20 mm2) detector installed on a Hitachi S3400N scanning electron microscope. Calibration of the instrument was performed on the Ti Kα at 4.509 keV. A powder X-ray diffraction (XRD) study was performed on a Bruker D8 Advance diffractometer using Cu-Kα radiation (λ = 1.5418 Å) at 45 kV and 40 mA, 0.05° step size and 60 s step time over a range of 1° to 80°. Small angle X-ray scattering (SAXS) measurements were performed on a pinhole type Rigaku Nanoviewer instrument using Cu-Kα radiation equipped with a CCD detector over a range of 1 to 5 degrees (2θ) respectively. The applied voltage and filament current were 40 kV and 50 mA respectively. The finely powdered samples were sealed in metal cells with polyethylene terephthalate films and kept in a temperature controlled sample chamber for measurements. Bragg spacing is calculated by d = 2π/q, where q is the magnitude of the scattering vector. Transmission electron microscopy (TEM) images were acquired with a FEI Tecnai 120 Twin microscope operating at 120 kV and equipped with a high resolution Gatan Orius CCD 4 k × 4 k numeric camera. The sample holder is equipped with an alpha-tilt (+70°/−70°) goniometer to observe the sample at different tilt angles and avoid 2D projections artefacts. Adsorption/desorption isotherms were recorded on a Belsorp–Max Instrument (BEL-Japan Inc.) using nitrogen as probe gas.Solid-state nuclear magnetic resonance (NMR) experiments were performed on a Bruker AVANCE III 300 (7.05 T) spectrometer using a wide-bore superconducting magnet. 1H and 27Al magic angle spinning (MAS) NMR spectra were recorded with a 4 mm MAS probe at a spinning frequency of 14 kHz. For the 1H and 27Al spectra, 32 and 4200 transients were collected with π/2 and π/12 pulses of 6.5 and 0.95 μs and at repetition times of 10 s and 500 ms respectively. The chemical shifts were referenced to TMS and Al(NO3)3 at 0 ppm respectively for 1H and 27Al. For the 13C{1H} cross-polarization (CP), one pulse 13C MAS, 29Si CP{1H} and one pulse 29Si MAS NMR studies, 7 mm zirconia rotors spinning at 5 kHz were employed. A total of 4096 transients were collected with a proton π/2 pulse of 6.5 μs and recycle delays of 1 s for the CP spectra of both nuclei. A cross-polarization contact time of 3 ms was applied for all the experiments. For 13C and 29Si MAS NMR experiments, 4096 and 1024 transients were collected with a π/4 pulse of 2.65 μs and at a relaxation delay of 10 and 60 s respectively. The chemical shift values were calibrated with reference to tetramethylsilane (TMS) at 0 ppm. The 1H{29Si{1H}} double CP were performed on a Bruker AVANCE III 300 (7.05 T) spectrometer. Details for double CP experiment are given in ref. 11. The 1H{27Al} CP experiments were performed on a Bruker AVANCE III 700 (16.45 T) spectrometer using a 3.2 mm rotor spinning at 23 kHz.
Results and discussion
Nanotube preparation
Imogolite was obtained by modification of earlier procedures,4,5 involving the mixing of acidic solutions of an aluminium salt with tetraethoxysilane, followed by neutralization and hydrothermal treatment. The main variations compared to the literature were the selection of aluminium chloride, the use of centimolar precursor concentration and the addition of glycerol α-monolaurate (MG).During washing procedures, we observed a different behavior for samples prepared with or without biosurfactant; whereas the purely inorganic preparation was easily dispersed in water, as expected for hydrophilic aluminosilicate nanotubes, the sample prepared with MG remained located at the air–water interface, suggesting that it exhibits a hydrophobic character. To confirm this hypothesis, 1-nonanol was added to the water solution. As shown in Fig. 1, after phase separation, the MG-free product was still in the bottom aqueous phase whereas the surfactant-templated sample was located in the top organic layer. This unexpected and unprecedented behavior of imogolite urged us to investigate in details the resulting materials first from an inorganic and organic point of view and then at the hybrid interface. Additional experiments led us to propose a reaction mechanism that is compatible with the one-step formation of hydrophobic imogolite nanotubes.
 | ||
| Fig. 1 Behaviour of aluminosilicate powders in a water/nonanol biphasic system: MG-free sample is easily dispersed in the bottom aqueous medium (left) whereas the MG-templated sample localizes in the top organic layer. | ||
Characterization of the inorganic structure
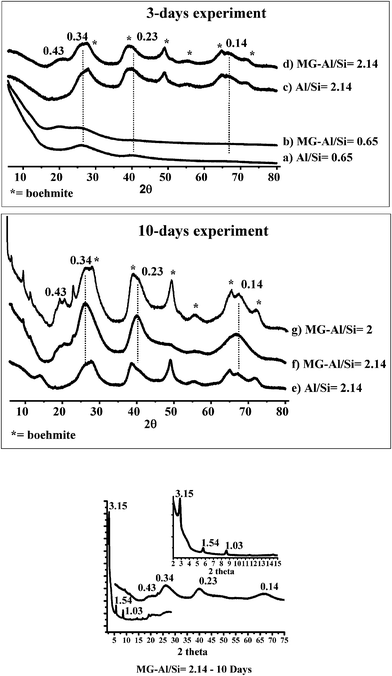 | ||
| Fig. 2 Powder X-ray patterns of MG (b, d, f, g) and MG-free (a, c, e) alumina-silica samples obtained after 3 days (a–d) and 10 days (e–g) aging time: (a, b) Al/Si = 0.65; (c, f) Al/Si = 2.14; (g) Al/Si = 2. Small and wide angle XRD of MG-Al/Si = 2.14 (10 days) sample is given in the inset. The given d-spacing values, in nm, and dotted lines refer to imogolite while asterisks indicate the presence of boehmite. | ||
The evolution from amorphous alumino-silicates to imogolite as a function of Al/Si molar ratio (3 days aging time) can also be followed by 29Si CP and 27Al solid-state MAS NMR experiments in Fig. 3. At low Al/Si values (3a, b), a broad peak centered at −93 ppm characterizes an amorphous alumino-silicate network made of Al sites, ranging from Q3(4Al) to Q4(4Al).13 At increasing Al3+ values (Fig. 3c, d), a characteristic peak at −78 ppm appears and, at Al/Si = 2.14 (Fig. 3d), it constitutes more than 30% of the overall silicon spectrum. This peak, in good agreement with previous studies,13a is extremely rare in aluminosilicates13b and is commonly assigned to (AlO)3SiOH species in the imogolite environment, that is silicon tetrahedron connected to a gibbsite layer.
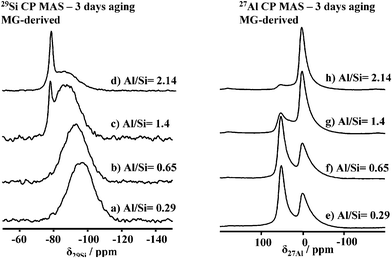 | ||
| Fig. 3 (a–d) 29Si CP and (e–h) 27Al MAS solid-state NMR experiments performed on MG-derived imogolite samples after 3 days aging and at different Al/Si ratios. | ||
A similar behavior occurs for the 27Al spectra evolution (Fig. 3e–h), where, at a low aluminum concentration (Fig. 3e–g), a mixture of octahedral (δ = 2.5 ± 0.5 ppm) and tetrahedral (δ= 55 ± 0.5 ppm) sites characterizes the material. At Al/Si values close to 2 (Fig. 3h), integration of the 27Al spectrum shows that more than 90% of the material is composed of octahedral aluminum sites, as expected for imogolite, allophanes and boehmite compounds.14 This is characteristic of the octahedral environment of aluminium polymerized in a gibbsite-like environment. A similar trend can be observed in MG-free samples (data not shown), showing that evolution from amorphous alumino-silicate to proto-imogolite does not actually depend on MG.
As mentioned earlier, the only way to distinguish between imogolite and proto-imogolite is either TEM or FT-IR spectroscopy. TEM images (Fig. 4a, b) show that MG-derived powder is composed of a bundle of fibers whose characteristic dimensions, length L > 50 nm and diameter d = 2.5 nm, confirm them as imogolite nanotubes. The SAED pattern extracted from Fig. 4a is given in Fig. 4c. The d-spacings calculated from the analysis of the diffraction rings numbered from (1) to (4) provide the following values: d(1) = 0.32 nm, d(2) = 0.21 nm, d(3) = 0.18 nm, d(4) = 0.14 nm. d(1), d(2) and d(4) can be attributed to the (071), (004) and (006) (hkl) diffraction planes observed for imogolite.5a On the contrary, the d(3) = 0.18 nm value corresponds to the (200) plane of the boehmite impurity, as discussed before.
 | ||
| Fig. 4 (a, b) Transmission electron microscopy images obtained for MG-derived imogolite (tubular morphology); c) SAED pattern obtained on image displayed in (a); (d) MG-free proto-imogolite (particle morphology). The Al/Si ratio is 2.14 and aging time is 3 days for all samples. | ||
MG-free samples (Fig. 4d) only show agglomerates of particles whose morphology is not fibrous. Therefore, at a Al/Si = 2.14 ratio, the presence of MG favors the formation of imogolite nanotubes within a 3 days aging period. However, the absence of significant peaks in the low-angle domain of the XRD pattern suggests that the nanotubes are completely disordered with no bundle organization at this stage.
Before proceeding with a deeper analysis of the 10 days samples, we observe that the amount of residual boehmite is particularly low in this sample (Fig. 2f), showing that our approach may lead to a rather pure imogolite provided that the synthetic conditions are optimized through a more systematic study. We did not explore this aspect any further in this work.
Very interestingly, nitrogen adsorption/desorption isotherms are of particular use to exclude the presence of a mesostructure lamellar solid. As shown in Fig. 1 in the supplementary information, the MG-Al/Si = 2.14—10 days sample, displays combined micro and mesoporosity, as indicated, respectively, by the nitrogen uptake below P/P0 = 0.2 and above P/P0 = 0.5, where a hysteresis loop occurs. In particular, after calcination at 350 °C, the amount of microporosity increases while the hysteresis loop remains intact. The presence of a hysteresis loop for the MG-containing and calcined sample excludes the presence of a lamellar structure. In fact, mesostructured, surfactant-containing, materials do not generally display any porosity and, most importantly, the lamellar framework collapses after calcination, which is not the case here. Any additional hypothesis of a layered aluminosilicate is also excluded by the sharp 29Si NMR peak at δ = −78 ppm, which cannot be attributed to any flat aluminosilicate surface.13b
Transmission electron microscopy images obtained for the MG and MG-free samples after 10 days are shown in Fig. 5(a–d). Evolution with time of the MG-free sample goes from morphologically undefined proto-imogolite (3 days aging, Fig. 4d) to disordered imogolite nanotubes (10 days aging, Fig. 5d). MG-containing samples, on the contrary, show imogolite nanotubes already at 3 days aging (Fig. 4a, b) and an interesting planar lamellar packing after 10-days aging, as shown on several sections of the samples in Fig. 5a–c. Throughout the sample, a large number of apparent sheets are actually composed of individual nanotubes arranged together in a parallel fashion. No three-dimensional order seems to exist between the nanotube sheets but superposition occurs in a rather unorganized way. Further confirmation of the existence of the nanotubes can be found in Fig. 5e–f, where we show an image of the same section, containing both individual nanotubes and sheets, before and after a 20° tilting of the sample holder. This manipulation allows one to discriminate between a true, uni-dimensional, nanotube and a side viewed, two-dimensional, platelet, which could actually be confused due to the projection of a 3D object on a two-dimensional surface. The black arrows point towards individual nanotubes before and after tilting and, as one can see, the indicated one does not change in lateral size upon tilting. This is of course expected for a nanotube but not for a platelet. TEM observations perfectly corroborate low-angle peaks in the XRD shown in Fig. 2 and are now attributed to a long-range order of the imogolite nanotubes within a single sheet.
 | ||
| Fig. 5 Transmission electron microscopy images obtained for (a–c) MG and (d) MG-free imogolite samples upon 10 days aging at Al/Si = 2.14. (e–f) Highlight on images observed at two different tilt angles of the sample holder with respect to the electron beam z-direction: 0° for (e) and +20° for (f). Arrows in both figures put in evidence the individual nanotubes before and after tilting showing no change in their thickness. | ||
This specific arrangement was never reported for imogolite and, at the moment, we can qualitatively attribute it to the presence of the MG on the surface of the tubes, as shown hereafter. Non-modified imogolite bundles are known to pack in a monoclinic crystal structure5 but, unfortunately, we could not find any report describing the packing of surface-modified imogolite. Park et al.5i have reported the two-dimensional alignment of octadecylphosphonic acid modified imogolite but that was only possible by using a post-synthesis modification of the fresh imogolite combined with a Langmuir–Blodgett technique. Nevertheless, despite the lack of experimental details, they briefly comment on the different packing of pure imogolite in bulk with modified imogolite nanotubes aligned on the surface as observed by XRD.
The faster formation of functionalized nanotubes in 2D lamellar packing in the presence of MG is highly reproducible and also occurs at the stoichiometric Al/Si = 2 ratio. In this situation, the final material after 10 days aging shows a good quality XRD pattern (Fig. 2g), typical for imogolite at wide angles, and of a lamellar phase, as described above, at low angles.
As a complementary source of information, the Al/Si ratio of the samples was measured by SEM/EDAX and averaged on different sections of the material. The experimental values were very similar to the initial stoichiometric content, i.e. 2.1 ± 0.1 when MG was present. In contrast, for an initial Al excess (Al/Si = 2.14), MG-containing samples showed a lower value (1.7 ± 0.1) whereas MG-free powders contain a more important Al excess (2.5 ± 0.1). These discrepancies (about 20%) could probably be justified by the presence of impurities in the final sample.
Characterization of the organic component
XRD and TEM have so far shown the important role of MG in favouring the formation of imogolite over proto-imogolite phase. In these conditions, TGA analyses (not shown) also revealed that the organic fraction constitutes between 20 and 30 wt% of the final materials. To understand this process better, we used a combination of multinuclear solid-state NMR experiments. Fig. 6 shows the 13C CP solid-state NMR spectra of MG-derived imogolite samples after 3 (Fig. 6b) and 10 (Fig. 6c) days aging. In a classical imogolite synthesis, one would not expect the presence of any major carbon species; here, both samples show the classical fingerprint of the carbon backbone of MG, as compared to the 13C solution spectrum of this compound (Fig. 6a).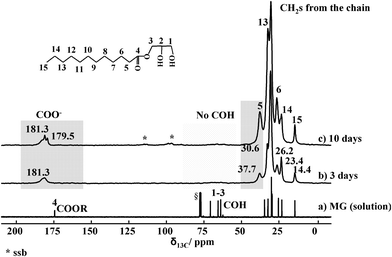 | ||
| Fig. 6 13C solution NMR spectrum of MG (a) and 13C CP MAS NMR spectra of MG-derived imogolite upon 3 (b) and 10 days (c) aging. Highlighted regions are discussed in the text. § refers to CDCl3. *: Spinning side bands. | ||
A close comparison between the solution and the solid-state spectra depicts some important differences:
(i) The glycerol moiety (between 60 and 75 ppm) of MG is lost during the synthetic process, as indicated by the lack of peaks 1–3 in the solid-state spectra (Fig. 6b, c). This is probably due to the hydrolysis of the ester bond between lauric acid and glycerol due to the acidic conditions used at the stage of AlCl3 addition.
(ii) The ester group in MG has a typical chemical shift value of δ = 175 ppm (Fig. 6a). The chemical shift of the same group in the imogolite samples is now around δ = 180 ppm, suggesting that the ester has been hydrolyzed into carboxylate groups COO−. This might result from the neutral pH conditions used for TEOS condensation and/or from complexation with Al3+ ions.
(iii) The chemical shift of peak 5, corresponding to the α-CH2 with respect to the ester function, shifts from 34 ppm (Fig. 6a) to 37 ppm (Fig. 6b, c), which may be due to the change in the chemical surrounding occurring on the carbonyl group. For instance, values at 37 ppm were reported for the α-CH2 group of aliphatic esters.15 Additionally, the relative intensities of peaks 5, 13 and 6 are affected at various aging times in imogolite. It was previously reported that adsorption of SDS on alumina provokes relative intensity variations with increasing SDS, but the authors could not provide a clear explanation.16 This point is still to be clarified.
Investigation of the hybrid interface
13C CP MAS NMR experiments show that MG undergoes hydrolysis during the material forming process. Glycerol, being water-soluble, is washed away and the remaining fatty acid alone (lauric acid) in its basic form is retained in the final material. Possible conformational changes and the unshielded value δ = 180 ppm for the COO−group suggest that MG lays on the nanotube surface. This idea is confirmed by 29Si- or 27Al-filtered 1H NMR solid-state experiments using both 1H{29Si{1H}} double cross polarization and 1H{27Al} cross polarization. The double cross polarization sequence uses the proton bath to polarize all atoms through an initial CP step at an optimized contact time, tCP1; then, 29Si magnetization is transferred to its nearest proton neighbors by employing a second, variable, contact time, tCP2. The shorter the tCP2 value, the closer the 29Si-to-proton distance is actually explored.Fig. 7a shows the single-pulse and 1H{29Si{1H}}double-CP experiments on the MG-derived sample after 3 days aging time. In the single pulse experiment, the methylene (CH2)x chain and the CH3 groups are detected between 0.5 and 2 ppm while H-bonded H2O is observed at 5 ppm. For double-CP experiments, the 29Si{1H} contact time (tCP1) is set to 3 ms while the second contact time (tCP2) varies between 500 μs and 10 ms. These values are optimized to explore both close and large internuclear distances. At all values of tCP2, only the H-bonded water (5 ppm) is detected while no MG-related signal is observed. The fatty acid is therefore not in close proximity with silicon, which excludes the direct functionalization of silicate layers of imogolite. However if an additional 10 ms delay time is set after cross-polarization, favoring uncontrolled long-range spin–spin interactions, the CH2 backbone signal is observed. The residual fatty acid is therefore intimately mixed with the imogolite sample and it is not phase separated.
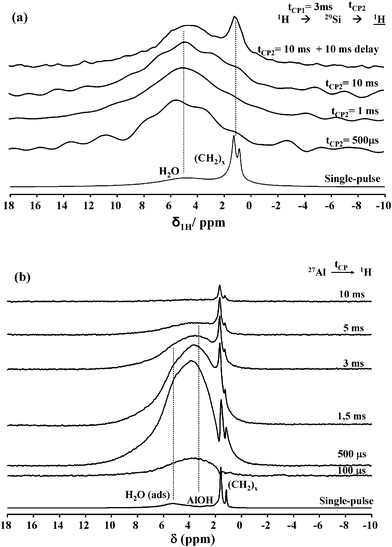 | ||
| Fig. 7 (a) 1H{29Si{1H}} double CP MAS NMR (B0 = 7.05 T; νMAS = 14 kHz) spectra of MG-derived imogolite upon 3 days aging time. tCP1 = 3 ms while tCP2 varies as indicated next to each spectrum. (b) 1H{27Al} CP MAS NMR (B0 = 16.45 T; νMAS = 23 kHz) spectra of MG-derived imogolite upon 3 days aging time. Contact times are provided next to each spectrum. The corresponding single pulse 1H MAS NMR spectra, recorded under comparable conditions (B0 and νMAS) for experiments shown in (a) and (b), are shown at the bottom of each set of experiments. | ||
As a complementary experiment, the proton signal was also detected after a 1H{27Al} cross polarization step. 1H{27Al} CP experiments provide a clear-cut answer on the localization of the residual fatty acid. The proton signal related to the fatty acid backbone (Fig. 7b) appears at tCP2 = 500 μs and its intensity increases continuously up to 10 ms. Additionally, this experiment partially filters the SiOH signal with respect to AlOH, δ = 3.6 ppm. The interpretation of these experiments is quite straightforward: the fatty acid is very close to the Al3+ centre, undoubtedly chelated by the carboxylate group (that cannot be detected directly via1H NMR experiments). These experiments are consistent with the specific nanotube morphology and structure parameters of imogolite, whose silicate layer is localized inside the tube while the gibbsite sheet is outside. Chemical and steric considerations help understanding that the C12fatty acid preferentially complexes aluminium atoms on the nanotube external surface rather than the inner silicon atoms. Indeed, the inner tube diameter is only 1 nm, thus smaller than the theoretical length (1.6 nm) of the lauric acid molecule in its all-trans configuration.17 All together, these data suggest that laurate residues functionalize the external surface of the nanotubes, providing a hydrophobic character to the whole sample, in agreement with our initial observations (Fig. 1).
Insights in the reaction mechanisms
Previous experiments show that MG hydrolyzes during the reaction and its laurate moiety finally ends as a complexing agent for Al3+ sites. In order to verify the role of glycerolversuslaurate in the formation mechanism, we have performed three test experiments with pure lauric acid, pure glycerol and a mixture of lauric acid and glycerol at 10 days aging time.Fig. 8 shows the wide angle X-ray diffraction patterns obtained for these samples: both samples containing lauric acid(b, c) have the typical pattern of imogolite nanotubes, whose packing is, once again, constituted of a 2D arrangement of nanotubes as shown by the diffractions peaks below 2θ = 15° and where the inter-tube distance, d, corresponds to 3.03 nm. On the contrary, no evidence of stacked imogolite nanotubes is obtained when glycerol is used alone (Fig. 8a). These data are confirmed by the TEM image of the lauric acid-derived sample in Fig. 8, showing a long-range order nanotube packing with an inter-tube distance of d = 3.2 nm, consistent with XRD data. These experiments support previous data on the MG system and indicate that lauric acid plays a key role in the formation of imogolite.
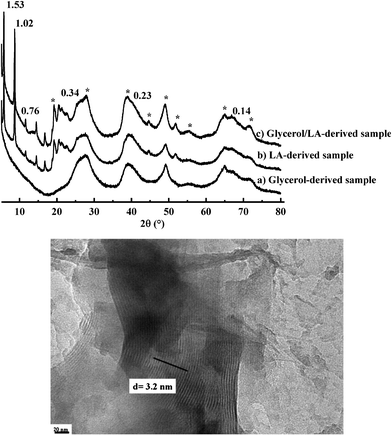 | ||
| Fig. 8 Top image: Wide angle X-ray diffraction patterns of (a) glycerol, (b) lauric acid (LA) and (c) glycerol/LA derived samples obtained after 10 days aging time (asterisks indicate the peaks due to boehmite structure). Values are given in nm and correspond to associated d-spacings. Bottom image: TEM image of lauric acid-derived imogolite at 10 days aging time. | ||
The synthesis of imogolite nanotubes has been reported so far under several conditions.4,5 In general, good quality imogolite can be obtained at millimolar Al3+ concentrations in the presence of TEOS as a silica source, two factors that contribute to favor hetero- (formation of Al–O–Si bonds) with respect to homo-condensation (formation of Al–O–Al and Si–O–Si bonds). Functionalization of the imogolite outer surface could only be achieved by post-synthesis functionalization with phosphonic acids. In our system, we combine a relatively high Al3+ concentration probably due to the stabilizing action of the surfactant. In a second stage, when proto-imogolite forms, laurate strongly interacts with gibbsite sheets, thus favoring the formation of curved surfaces, already imposed by the silicate tetrahedra.5a,18Laurate-functionalized imogolite nanotubes are then formed. Finally, side-by-side interactions between nanotubes are assisted by van der Waals interactions between laurate alkyl chains, that favor nanotube packing. At the moment, even if we do not have a clear explanation of the reason why packing occurs only within a plane and not in the 3rd dimension, providing so-called “raft-like” objects, we can nevertheless formulate some hypothesis.
In the domain of carbon nanotubes, it was shown that packing (hexagonal vs. columnar-like) can be influenced by their chemical surrounding.19,20 In particular, Jeong et al.20 have shown that single-walled carbon nanotubes can pack in two-dimensional plate-like sheets using a smectic liquid crystal template. In our system, the amount of MG is not enough to justify the formation of a liquid crystal. Nevertheless, in the field of surfactant-templated mesostructured silica and aluminosilica materials, the formation of lyotropic hybrid organic/inorganic phases occurs both under a liquid crystal like approach (high concentration of surfactant)21 and under dilute conditions, where self-organisation and precipitation is driven in solution by the cooperative formation of inorganic–organic interfaces, as proposed by Monnier et al.22 Chmelka and co-workers also reported on the formation of mesostructured aluminosilicate by the same approach and, interestingly, their advanced solid state NMR study showed that incorporation of aluminum in the inorganic framework did not induce a 29Si chemical shift at −78 ppm,23 which supports the fact that in our case the formation of a classical mesoporous aluminosilicate is most-likely excluded.
Plate, “raft” and nanosheets (nomenclature varies according to the authors) aluminosilicate materials have been described earlier. Barret24 and Zhao,25 for instance, reported, respectively, about the formation of pure and Ga-doped boehmite “rafts”. Nevertheless, in their case, the “raft” population was described as being uniform in size and composed of either “units” or layered materials; in particular, XRD did not show any small-angle reflection at all, excluding any long-range order in the mesoscale range. In our work, the 2D plates are not homogeneous in size and are composed of nanotubes of different lengths, as clearly shown by TEM experiments at different tilt angles (Fig. 5e, f). Interestingly, “raft”-like objects have been reported for imogolite before,26 and in particular several authors have pointed out the liquid crystal behavior of imogolite nanotubes at a high concentration, as predicted by the Onsager and Flory theory.26c
In our case, we think that the plate-like sheets composed of imogolite nanotubes are driven by a synergic interaction between the laurate and the imogolite nanotubes and assembly may occur during the drying process at the liquid/air interface, as predicted by several authors for these types of objects.27Scheme 1 proposes a picture summarizing the main achievement of this work. In order to describe properly the sharp small-angle reflections in the XRD pattern, one has to assume that each plate contains a large number of nanotube units. Finally, uncontrolled side reactions, probably due to non-optimized MG/Al/Si stoichiometry, lead to the formation of boehmite.
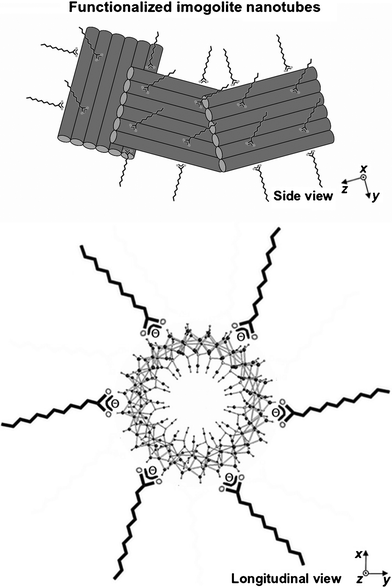 | ||
| Scheme 1 Schematic side and longitudinal tentative representation of the hydrophobic imogolite nanotubes. The outer surface of the tube is composed of gibbsite sheets whose Al3+ sites are complexed by lauric acid. The longitudinal view is adapted from ref. 5e. The size of the plates in this drawing is not to scale. In particular one has to consider the presence of a large number of nanotubes in order to account for the sharp XRD reflections observed in the small-angle range. | ||
Conclusions
Among inorganic nanotubes, imogolite represents a very unique case due to its natural occurrence. Therefore, in contrast to other metal oxide phases whose morphology and dimensions can be controlled by templating approaches,28 the addition of organics is not expected to change the structure of individual nanotubes but may control their kinetics of formation and their packing, as demonstrated here. Of particular interest is the possibility to achieve functionalization of the imogolite surface in one-step and achieve a new type of nanotube arrangement, never observed before for imogolite; additionally, this is done in a short time and starting from a high inorganic precursor concentration. This was possible by the selection of a surfactant molecule that exhibits a good affinity for the aluminium species, both in the initial precursor solution and within the aluminosilicate framework. An additional, unexpected outcome of this specific affinity is that a direct surface functionalization of the nanotube is occurring, turning the hydrophilic aluminosilicate particles into a solvent-dispersable powder. Together with the fact that our synthetic pathway complies with several key requirements of green chemistry principles applied to inorganic materials (solvent-free, near neutral pH, biosurfactant, use of chloride instead of perchlorate aluminium salt,..),29 it can be expected that these results will open new perspectives for the use of imogolite as a functional filler in the field of (nano)-composite materials.30Acknowledgements
B. T. thanks the Conseil Régional d'Ile-de-France for his post-doctoral fellowship (Allocations post-doctorales Hors DIM “Chimie Verte et Eco-matériaux”). M. Selmane, G. Mosser and P. Le Griel (LCMCP, Paris, France) are acknowledged for their help in XRD, SAXS and TEM experiments. We thank Prof. J. P. Jolivet, C. Chanéac (LCMCP, Paris, France) and Dr C. Levard (CEREGE, Aix-Marseille, Paris, France) for enlightening discussions. This project was supported by an AIC CPDD-RDR1 from CNRS/INRA. We thank reviewer one for the helpful comments.References
- (a) P. Avouris, Acc. Chem. Res., 2002, 35, 1026 CrossRef CAS; (b) C. N. R. Rao and A. Govindaraj, Adv. Mater., 2009, 21, 4208 CrossRef CAS; (c) W. C. Ackerman, D. M. Smith, J. C. Huling, Y. W. Kim, J. K. Bailey and C. J. Brinker, Langmuir, 1993, 9, 1051 CrossRef CAS; (d) S. J. Son, X. Bai and S. B. Lee, Drug Discovery Today, 2007, 12, 650 CrossRef CAS; (e) J. Goldberger, R. Fan and P. D. Yang, Acc. Chem. Res., 2006, 39, 239 CrossRef CAS; (f) F. Y. Cheng and J. Chen, J. Mater. Res., 2006, 21, 2744 CrossRef CAS; (g) J. B. Harsh, S. J. Train, J. Boyle and Y. Tang, Clays Clay Miner., 1992, 40, 700 CAS; (h) K. Balasubramanian and M. Burghard, Anal. Bioanal. Chem., 2006, 385, 452 CrossRef CAS; (i) Y. Arai, M. McBeath, J. R. Bargar, J. Joye and J. A. Davis, Geochim. Cosmochim. Acta, 2006, 70, 2492 CrossRef CAS; (j) S. Imamura, Y. Hayashi, K. Kajiwara, H. Hoshino and C. Kaito, Ind. Eng. Chem. Res., 1993, 32, 600 CrossRef CAS; (k) K. Yamamoto, H. Otsuka and A. Takahara, Polym. J., 2007, 39, 1 CrossRef CAS.
- (a) M. S. Dresselhaus and H. Dai, MRS Bull., 2004, 29, 237 CrossRef; (b) W. A. de Heer, MRS Bull., 2004, 29, 281 CrossRef CAS; (c) R. Tenne and G. Seifert, Annu. Rev. Mater. Res., 2009, 39, 387 CrossRef CAS.
- (a) K. Wada and N. Yoshinga, Am. Mineral., 1969, 54, 50 CAS; (b) P. D. Cradwick, K. Wada, J. D. Russell, N. Yoshinaga, C. R. Masson and V. C. Farmer, Nature, 1972, 240, 187 CrossRef CAS; (c) V. C. Farmer, A. R. Fraser and J. M. Tait, J. Chem. Soc., Chem. Commun., 1977, 462 RSC; (d) V. C. Farmer, B. F. L. Smith and J. M. Tait, Clay Miner., 1979, 14, 103 CAS; (e) S. I. Wada, A. Eto and K. Wada, J. Soil Sci., 1979, 30, 347 CrossRef CAS; (f) V. C. Farmer, M. J. Adams, A. R. Fraser and F. Palmieri, Clay Miner., 1983, 18, 459 CAS; (g) M. A. Wilson, N. H. Tran, A. S. Milev, G. S. K. Kannangara, H. Volk and G. Q. M. Lu, Geoderma, 2008, 146, 291 CrossRef CAS; (h) K. Wada, Dev. Sedimentol., 1978, 26, 147 CrossRef CAS.
- (a) K. Inoue and P. M. Huang, Nature, 1984, 308, 58 CrossRef CAS; (b) K. Inoue and P. M. Huang, Clays Clay Miner., 1985, 33, 312 CAS; (c) S. M. Barret, P. M. Budd and C. Price, Eur. Polym. J., 1991, 27, 609 CrossRef; (d) G. H. Koenderink, S. G. J. M. Kluijtmans and A. P. Philipse, J. Colloid Interface Sci., 1999, 216, 429 CrossRef CAS.
- (a) S. Mukherjee, M. Veda, V. M. Bartlow and S. Nair, Chem. Mater., 2005, 17, 4900 CrossRef CAS; (b) S. Konduri, S. Mukherjee and S. Nair, ACS Nano, 2007, 1, 393 CrossRef CAS; (c) C. Levard, A. Masion J. Rose, E. Doelsch, D. Borschneck, C. Dominici, F. Ziarelli and J. Y. Bottero, J. Am. Chem. Soc., 2009, 131, 17080 CrossRef CAS; (d) H. Yang, C. Wang and Z. Su, Chem. Mater., 2008, 20, 4484 CrossRef CAS; (e) B. Bonelli, I. Bottero, N. Ballarini, S. Passeri, F. Cavani and E. Garrone, J. Catal., 2009, 264, 15 CrossRef CAS; (f) I. Bottero, B. Bonelli, S. E. Ashbrook, P. A. Wright, W. Zhou, M. Tagliabue, M. Armandi and E. Garrone, Phys. Chem. Chem. Phys., 2011, 13, 744 RSC; (g) D.-Y. Kang, J. Zang, E. R. Wright, A. L. McCanna, C. W. Jones and S. Nair, ACS Nano, 2010, 4, 4897 CrossRef CAS; (h) K. Yamamoto, H. Otsuka, S.-I. Wada, D. Sohn and A. Takahara, Polymer, 2005, 46, 12386–12392 CrossRef CAS; (i) S. Park, Y. Lee, B. Kim, J. Lee, Y. Jeong, J. Noh, A. Takahara and D. Sohn, Chem. Commun., 2007, 2917–2919 RSC.
- (a) Y. Q. Zhu, W. K. Hsu, H. Terrones, N. Grobert, B. H. Chang, M. Terrones, B. Q. Wei, H. W. Kroto, D. R. M. Walton, C. B. Boothroyd, I. Kinloch, G. Z. Chen, A. H. Windle and D. J. Fray, J. Mater. Chem., 2000, 10, 2570 RSC; (b) R. B. Little, J. Cluster Sci., 2003, 14, 135 CrossRef CAS; (c) M. Remskar, Adv. Mater., 2004, 16, 1497 CrossRef CAS; (d) R. E. Cochran, J. J. Shye and N. P. Padture, Acta Mater., 2007, 55, 3007 CrossRef CAS.
- (a) E. J. Strandine, G. T. Carlin, G. A. Werner and R. P. Hopper, Cereal Chem., 1951, 28, 449 CAS; (b) R. B. R. Choudhury, J. Am. Oil Chem. Soc., 1962, 39, 345 CrossRef; (c) J. B. Lauridsen, J. Am. Oil Chem. Soc., 1976, 53, 400 CrossRef CAS.
- (a) N. J. Krog, In Food emulsifiers and their chemical and physical properties, S. E. Friberg and K. Larson (eds), Marcel Dekker, New York, 1997, pp. 141-188 Search PubMed; (b) L. Sagalowicz, M. E. Leser, H. J. Watzke and M. Michel, Trends Food Sci. Technol., 2006, 17, 204 CrossRef CAS.
- (a) Z. N. Atay and B. H. Robinson, Langmuir, 1999, 15, 5056 CrossRef; (b) D. Chiche, C. Chizallet, O. Durupthy, C. Chanéac, R. Revel, P. Raybaud and J.-P. Jolivet, Phys. Chem. Chem. Phys., 2009, 11, 11310 RSC; (c) Z. Bachar, Z. Mouloungui and V. Noirot, Bioinorg Chem. Appl., 2010 Search PubMed , Article ID 292760.
- Z. Mouloungui and C. Gauvrit, Ind. Crops Prod., 1998, 8, 1 CrossRef CAS.
- N. Baccile, G. Laurent, C. Bonhomme, P. Innocenzi and F. Babonneau, Chem. Mater., 2007, 19, 1343 CrossRef CAS.
- G. G. Christophe, C. E. Corbato, D. A. Hofmann and R. T. Tettenhorst, Clays Clay Miner., 1979, 27, 81 Search PubMed.
- (a) P. F. Barron, M. A. Wilson, A. S. Campbell and R. L. Frost, Nature, 1982, 299, 616 CrossRef CAS; (b) E. E. Lippmaa, M. Magi, A. Samoson, G. Engelhardt and A. R. Grimmer, J. Am. Chem. Soc., 1980, 102, 4889 CrossRef CAS; (c) C. I. Merzbacher, K. J. McGrath and P. L. Higby, J. Non-Cryst. Solids, 1991, 136, 249 CrossRef CAS.
- (a) S. Hiradate, H. Hirai and H. Hashimoto, Geoderma, 2006, 136, 696 CrossRef CAS; (b) R. A. Kinsey, R. J. Kirkpatrick, J. Hower, K. A. Smith and E. Oldfield, Am. Mineral., 1985, 70, 537 CAS; (c) P. Ildefonse, R. J. Kirkpatrick, B. Montez, G. Calas, A. M. Flank and P. Lagarde, Clays Clay Miner., 1994, 42, 276 CAS.
- M. G. Vachon and J. G. Nairn, Eur. J. Pharm. Biopharm., 1998, 45, 9 CrossRef CAS.
- G. Piedra, J. J. Fitzgerald, C. F. Ridenour and G. E. Maciel, Langmuir, 1996, 12, 1958 CrossRef CAS.
- H. N. Patrick, G. G. Warr, S. Manne and I. A. Aksay, Langmuir, 1999, 15, 1685 CrossRef CAS.
- S. Mukherjee, K. Kim and S. Nair, J. Am. Chem. Soc., 2007, 129, 6820 CrossRef CAS.
- D. J. Needleman, M. A. Ojeda-Lopez, U. Raviv, H. P. Miller, L. Wilson and C. R. Safinya, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16099–16103 CrossRef CAS.
- H. S. Jeong, Y. K. Ko, Y. H. Kim, D. K. Yoon and H.-T. Jung, Carbon, 2010, 48, 774–780 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- A. Monnier, F. Schueth, Q. Huo, D. Kumar, D. Margolese, R. S. Maxwell, G. D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B. F. Chmelka, Science, 1993, 261, 1299 CAS.
- M. T. Janicke, C. C. Landry, S. C. Christiansen, D. Kumar, G. D. Stucky and B. F. Chmelka, J. Am. Chem. Soc., 1998, 120, 6940–6951 CrossRef CAS.
- S. M. Barret, P. M. Budd and C. Price, Eur. Polym. J., 1991, 27, 609 CrossRef.
- Y. Zhao, R. L. Frost and W. N. Martens, J. Phys. Chem. C, 2007, 111, 5313 CAS.
- (a) A. S. Sonin, J. Mater. Chem., 1998, 8, 2557–2574 RSC; (b) K. Kajiwara, H. Urakawa, N. Donkai, Y. Hiragi, H. Inagaki and M. Schmidt, Bull. Inst. Chem. Res., Kyoto Univ., 1988, 66(3), 165 CAS; (c) K. Kajiwara, N. Donkai, Y. Hiragi and H. Inagaki, Makromol. Chem., 1986, 187, 2895 CrossRef CAS; (d) K. Kajiwara, N. Donkai, Y. Fujiyoshi, Y. Hiraki, H. Urakawa and H. Inagaki, Bull. Inst. Chem. Res., Kyoto Univ., 1985, 63(34), 320 CAS; (e) H. Hoshino, H. Urakawa, N. Donkai and K. Kajiwara, Polym. Bull., 1996, 36, 257–264 CrossRef CAS.
- A. Marmur and R. H. A. Ras, Soft Matter, 2011, 7, 7382 RSC.
- (a) H. J. Muhr, F. Krumeich, U. P. Schönholzer, F. Bieri, M. Niederberger, L. J. Gauckler and R. Nesper, Adv. Mater., 2000, 12, 231 CrossRef CAS; (b) S. Kobayashi, N. Hamasaki, M. Suzuki, M. Kimura, H. Shirai and K. Hanabusa, J. Am. Chem. Soc., 2002, 124, 6550 CrossRef CAS; (c) J. H. Jung, S.-H. Lee, J. S. Yoo, K. Yoshida, T. Shimizu and S. Shinkai, Chem.–Eur. J., 2003, 9, 5307 CrossRef CAS.
- N. Baccile, F. Babonneau, B. Thomas and T. Coradin, J. Mater. Chem., 2009, 19, 8537 RSC.
- (a) W. Ma, H. Otsuka and A. Takahara, Chem. Commun., 2011, 47, 5813 RSC; (b) K. Yamamoto, H. Otsuka, S.-I. Wada, D. Sohn and A. Takahara, Soft Matter, 2005, 1, 372 RSC.
| This journal is © The Royal Society of Chemistry 2012 |