Double-heterojunction structure of SbxSn1-xO2/TiO2/CdSe for efficient decomposition of gaseous 2-propanol under visible-light irradiation
Sher Bahadur
Rawal
,
Ashok Kumar
Chakraborty
,
Yong Joo
Kim
,
Hark Jin
Kim
and
Wan In
Lee
*
Department of Chemistry, Inha University, Incheon 402-751, Republic of Korea. E-mail: wanin@inha.ac.kr; Fax: +82-32-867-5604; Tel: +82-32-863-1026
First published on 15th November 2011
Abstract
Highly crystallized antimony-doped tin oxide (ATO; SbxSn1-xO2, x = 0.1) of ∼50 nm size was prepared by co-precipitation of SnCl4·5H2O and SbCl3, followed by heat-treatment at 1000 °C. The prepared ATO nanoparticles of deep blue color revealed a profound light-absorption in the visible range. ATO/TiO2 composites were prepared by covering the surface of ATO nanoparticles with TiO2 using the sol–gel method. Under visible-light irradiation (λ ≥ 420 nm), the prepared ATO/TiO2 showed a notable photocatalytic efficiency in decomposing gaseous 2-propanol (IP), which seemed to be caused by the hole-transfer mechanism between the valence bands (VB) of ATO and TiO2, since the ATO's VB level is located lower than that of TiO2. Subsequently, a double-heterojunction ATO/TiO2/CdSe structure was prepared by loading CdSe quantum dots (QDs) onto the surface of the ATO/TiO2, which dramatically enhanced the visible-light photocatalytic efficiency. In fact, the catalytic activity of ATO/TiO2/CdSe in evolving CO2 from IP, was ∼3 times that of ATO/TiO2 and twice that of typical N-doped TiO2. The unexpectedly high efficiency of ATO/TiO2/CdSe seemingly is due to the unique band matching among these semiconductors. With sensitization of ATO and CdSe, not only the holes but also the electrons are generated in the VB and CB, respectively, of TiO2 under visible-light irradiation.
1. Introduction
Photocatalytic decomposition of organic pollutants has attracted widespread attention over the last few decades. TiO2 is known as the most efficient photocatalyst among the various semiconductors, due to its unique characteristics in band position, surface structure, and electron mobility, as well as its extended chemical stability and non-toxicity.1–6 However, TiO2, due to its wide band gap (Eg = 3.2 eV), can utilize only the photons in the UV region (λ < 380 nm), which limits its commercial applications in sunlight or indoors.7–11 Hence, in order to be able to utilize the major portion of the solar spectrum, development of photocatalysts that are functional under visible-light is indispensable.Thus far, several design strategies for visible-light photocatalysts have been pursued, among which are lowering of the TiO2 conduction band (CB) level by doping transition metal ions,12–17 and elevation of the valence band (VB) level by substituting anions such as N, C, S, or B for the oxygen sites in TiO2.18–22 Recently, utilization of charge transfer reaction by grafting metal ions onto the TiO2 surface has been reported,23–25 and it seems to be a noticeable strategy in designing visible-light photocatalysts. Significant progress has been made thus far, but for the purpose of practical applications, the visible-light photocatalytic efficiency must be further improved.
Another promising approach is the formation of a heterojunction structure between the sensitizer and the main photocatalyst. According to the relative energy band location there, two kinds of heterojunctions can be designed. First, as outlined in Scheme 1(a) (the Type-A heterojunction), the CB of sensitizer semiconductor is positioned higher than that of the main photocatalyst, in the present case TiO2. The Type-A heterojunction can be formed by loading, for example, metal chalcogenide quantum dots (QDs) or inorganic dyes onto the TiO2 surface. Under visible-light irradiation, the sensitizer-A is excited, and the electrons are then transported to the TiO2 CB. These electrons can induce various reduction reactions and participate in decoloration reactions of organic dyes,26–30 but CO2 evolution resulting from complete oxidation of organic pollutants is difficult, due to the unavailability of the OH˙ radical on the TiO2 surface. Serpone et al. reported that complete decomposition also is possible with CdS/TiO2,31 but the efficiency is low, because the photocatalytic reaction occurs via the following complicated steps.
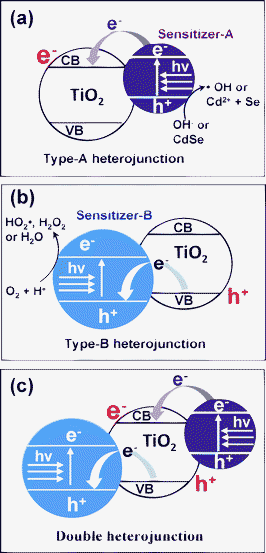 | ||
| Scheme 1 (a) Schematic diagram of Type-A heterojunction. Under visible-light irradiation, sensitizer-A is excited, and the photo-excited electrons are injected into the CB of TiO2. (b) Schematic diagram of Type-B heterojunction. Under visible-light irradiation, sensitizer-B is excited, and the induced holes are transported to the VB of TiO2. (c) Schematic diagram of double-heterojunction. Under visible-light irradiation, both the sensitizer-A and B are excited. As a result, electrons and holes are generated in the TiO2 CB and VB, respectively. | ||
| CdS + hν → CdS(e−CB + h+VB) | (1) |
| CdS(e−CB + h+VB) + TiO2 → CdS(h+VB) + TiO2(e−CB) | (2) |
| TiO2(e−CB) + O2 → TiO2 + O·−2 | (3) |
| O·−2 + H+ → HO2˙ | (4) |
| HO2˙ + Organic(ads) → Intermediates → Products | (5) |
VB position of SnO2 (+ 3.6 eV vs. NHE) has been reported to be considerably lower than that of TiO2 (+ 2.9 eV vs. NHE), but its wide bandgap (Eg), 3.5 eV, does not allow utilization of visible-light.31,38,39 In the present work, we doped Sb ions into SnO2 to form antimony-doped tin oxide (ATO), which absorbs a considerable portion of visible-light with its deep-blue color. We found that ATO is an effective sensitizer for use with TiO2 in forming the Type-B heterojunction structure with high visible-light photocatalytic activity.
For further enhancement of visible-light catalytic efficiency, we doubly combined the two different sensitizers with TiO2. That is, ATO (sensitizer-B) was designed to be located in the core of the TiO2 structure, whereas CdSe (sensitizer-A) was loaded onto the TiO2 surface. According to Scheme 1(c), both sensitizers are excited by visible-light irradiation, and resultantly the electrons and holes are transported to the CB and VB, respectively, of the TiO2. These electrons and holes generated on TiO2 are expected to enhance photocatalytic oxidation reactions, since not only the holes but also the electrons are considered to be essential for the efficiency of those oxidation reactions. In the present work, the photocatalytic behavior of ATO/TiO2/CdSe and the mechanistic roles of ATO and CdSe in enhancing its photocatalytic oxidation reactions were investigated.
2. Experimental section
2.1. Preparation of ATO
The ATO nanoparticles, used in forming the heterojunction with TiO2, were synthesized by the co-precipitation and subsequent heat-treatment.40–43 In a typical synthesis of 10 mol% Sb-doped ATO particles, 9 mmol of tin(IV) chloride pentahydrate (SnCl4·5H2O, Aldrich) and a stoichiometric amount of antimony(III) chloride (SbCl3, Aldrich) were dissolved in 50 ml of anhydrous ethanol. With vigorous stirring, tetrabutyl ammonium hydroxide (TBAH, 40 wt% water solution, Aldrich) was added dropwise, until the pH of solution was adjusted to 7.5. In the course of the stirring, Sn and Sb species were gradually co-precipitated, and the collected precipitate was washed with de-ionized water several times. It was dried in air at 60 °C for 12 h, and then calcined at 1000 °C for 2 h.2.2. Preparation of ATO/TiO2 composites
A total of 3.67 g titanium isopropoxide (97%, Aldrich) was stabilized in a mixed solution of 40 ml ethanol, 1 ml concentrated nitric acid, and 1 ml water. Then the prepared Ti-precursor solution was gently stirred for 4 h. A stoichiometric amount of 10 mol% Sb-doped ATO particles was then added to this solution. In order to obtain 5/95 ATO/TiO2 composite (in wt% ratio), 53 mg 10 mol% Sb-doped ATO particles pre-suspended in 10 ml ethanol was added and gently stirred overnight. The sample was then dried at 80 °C for 24 h, and subsequently heat-treated at 250 °C for 2 h. As a blank sample, the bare TiO2 was prepared by the same procedure, but without adding ATO.2.3. Preparation of cadmium selenide (CdSe) and its surface modification
Cadmium selenide (CdSe) quantum dots (QDs) were synthesized in noncoordinating solvent octadecene (ODE, Aldrich) with the addition of trioctylphosphine oxide (TOPO, Aldrich) as a capping agent.44 Typically, 0.0154 g CdO, 0.339 g oleic acid (OA), 2.72 g ODE, and 0.928 g TOPO were mixed in a three-neck flask, and heated to 300 °C under an anhydrous N2 flow. A solution consisting of 0.0190 g Se, 0.356 g trioctylphosphine (TOP, Aldrich) and 1.625 g ODE in a beaker was quickly added to the heated solution. The temperature of the mixture was then adjusted to 260 °C so as to induce the growth of CdSe nanocrystals. After 5 min, the reaction was quenched by adding 30 ml of cold toluene.The TOPO capping the CdSe QD was exchanged for mercaptopropionic acid (MPA, Aldrich) following the procedure reported by Leschkies et al.,45 in order to allow for CdSe QD attachment to the TiO2 surface. According to the typical procedure, 0.2 mmol MPA was dissolved in 100 mL anhydrous methanol, the pH of which solution was adjusted to 11.4 by adding tetramethylammonium hydroxide (TMAH, Aldrich). 40 mg TOPO-capped CdSe was then added this solution. The mixed solution was heated to 63 °C under Ar atmosphere, and refluxed for ∼24 h in darkness. The formed MPA-capped CdSe QD was precipitated by adding a mixture of ethyl acetate and diethyl ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume). The collected precipitate was washed several times with ethyl acetate to remove residual MPA or TOPO. The prepared CdSe is then readily soluble in protic solvents.
:1 in volume). The collected precipitate was washed several times with ethyl acetate to remove residual MPA or TOPO. The prepared CdSe is then readily soluble in protic solvents.
2.4. Preparation of ATO/TiO2/CdSe composites
Into a 50 ml ethanol suspension containing 1.00 g 5/95 ATO/TiO2 composite, stoichiometric amounts of the MPA-capped CdSe were dispersed, and the resultant solution was stirred at 60 °C for 6 h. In order to obtain, for example, 1 wt% CdSe-loaded ATO/TiO2 composite, 10.1 mg MPA-capped CdSe was added. It is known that during the stirring the MPA-capped CdSe QD can bind to the surface of the ATO/TiO2, due to the high binding affinity of the carboxylic group for TiO2. The formed ATO/TiO2/CdSe composites were then precipitated by centrifugation, and the collected particles were heated at 250 °C for 2 h.2.5. Characterizations
X-Ray diffraction (XRD) patterns of the prepared photocatalytic samples were obtained using a Rigaku Multiflex diffractometer with monochromatic light-intensity Cu Kα radiation. XRD scanning was performed under ambient conditions over 2θ region of 20–80° at a rate of 2°/min (40 kV, 20 mA). UV-visible diffuse reflectance spectra were acquired by a Perkin-Elmer Lambda 40 spectrophotometer. BaSO4 was used as the reflectance standard. Transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDX)-mapping images were obtained by a JEOL JEM2100F operated at 200 kV. One milligram of the synthesized particles was dispersed in 50 mL of ethanol, and a drop of the suspension was then spread on a holey amorphous carbon film deposited on the copper grid.2.6. Evaluation of photocatalytic activity
The visible-light photocatalytic efficiencies of the photocatalytic samples were estimated by monitoring the decomposition of gaseous 2-propanol (IP). An aqueous suspension containing 8.0 mg of the photocatalytic samples was spread on a 2.5 × 2.5 cm2 Pyrex glass in film form and subsequently dried at room temperature. Preparatory to the photocatalytic measurement, the prepared films were irradiated by a 300 W Xe lamp for 3 h, in order to remove possible organic residues. The gas reactor system used to measure the photocatalytic activity has been described elsewhere.46 The net volume of the gas-tight reactor was 200 mL, and the photocatalytic film was located at the center of the reactor. The entire area of the photocatalytic film (2.5 × 2.5 cm2) was irradiated by a 300 W Xe lamp through a UV cut-off filter (λ < 420 nm, Oriel) and a water filter to cut-off the IR spectrum. After evacuation of the reactor, 1.6 μL of the water-diluted IP (IP : H2O = 1:9 in volume) was injected into the reactor. The initial concentration of gaseous IP in the reactor was maintained at 117 ppm in volume (ppmv). Thus the ultimate concentration of CO2 evolved, with all of the IP decomposed, would be 351 ppmv, as shown in the following equation: | 2 (CH3)2CHOH (g) + 9 O2 (g) → 6 CO2 (g) + 8 H2O (g) | (6) |
The total pressure of the reactor was then increased to 750 Torr by the addition of oxygen gas. Under these conditions, the IP and H2O remained in the vapor phase. After a certain duration of irradiation, 0.5 mL of the gas in the reactor was automatically collected and sent to a gas chromatograph (Agilent Technologies, Model 6890N) using an auto sampling valve system. For CO2 detection, a methanizer was installed between the GC column outlet and the FID detector.
3. Results and discussion
ATO nanoparticles at different Sb-doping levels were prepared by co-precipitation and subsequent 2 h heat-treatment at 1000 °C. As the Sb-doping level was increased, the bluish color of the ATO particles grew bolder and deeper. This suggests that a higher doping level would induce more efficient utilization of visible light, but in fact it was found from the X-ray diffraction patterns shown in Fig. 1(a) that only a limited amount of Sb ion could be doped into the SnO2 lattice. That is, the pure cassiterite SnO2 phase (JCPDS, No. 77-0450) could be maintained only up to 10 mol% of Sb incorporation, whereas further incorporation led to the formation of impurity peaks, suggesting that extra Sb species were segregated to form the Sb2O4 cervantite phase (JCPDS, No. 77-0127). This is consistent with previous observations.47–49 The average crystallite size of the 10 mol% Sb-doped ATO particles, as determined from the (110) and (101) peaks by application of the Scherrer equation, was ∼39 nm, strongly indicating the highly crystallized structure.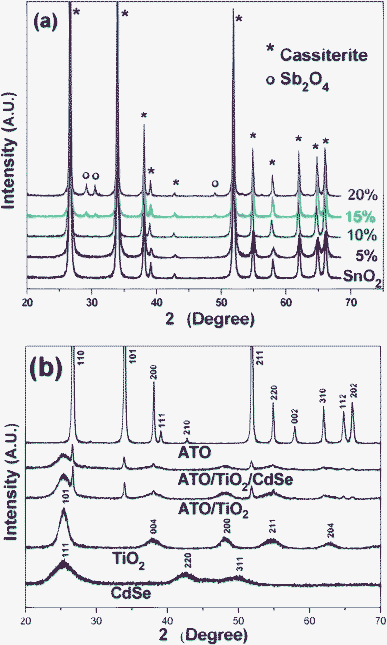 | ||
| Fig. 1 (a) XRD patterns of SnO2 and several ATO nanoparticles of different compositions. (b) XRD patterns of CdSe, TiO2, ATO (SbxSn1-xO2, x = 0.1), 5/95 ATO/TiO2, and ATO/TiO2/CdSe (CdSe: 1 wt%) composites. | ||
Fig. 2(a) shows the 10 mol% Sb-doped ATO powder exhibiting a deep blue color. This color is associated with ATO's internal energy bands formed between the CB and VB of SnO2. In the ATO lattice, the oxidation state of the Sb ions substituting for Sn4+ will be 5+ and 3+. Hence, its chemical formula can be written Sn4+0.9Sb5+xSb3+0.1-xO1.95+x, where 0 < x < 0.1.50 The presence of Sb5+ and Sb3+ ions produces additional energy bands originating from Sb 4d and 5p, below the SnO2 CB. Thus the CB level is lowered considerably, whereas the VB level originating from O 2p is not appreciably affected. As a result, the bandgap of ATO is reported to be 2.55 eV or less,51–53 which allows for visible light absorption.
 | ||
| Fig. 2 (a) Photographic image of ATO (SbxSn1-xO2, x = 0.1) powders. (b) TEM image of ATO nanoparticles. (c) High-resolution TEM image of a single ATO nanoparticle. (d) Magnification of dotted-rectangular area indicated in (c). (e) SAED pattern obtained from entire area of single ATO nanoparticle is shown in (c). | ||
Fig. 2(b) and 2(c) show TEM images of 10 mol% Sb-doped ATO particles of ∼50 nm size. The particles are moderately monodispersed and mostly mutually separated, without significant agglomeration. The dotted rectangular area of the single ATO particle shown in Fig. 2(c) was further magnified (Fig. 2(d)). The TEM image reveals the fringe patterns uniformly formed over the entire area, suggesting that each ATO particle is a single crystal. The spacing of the fringe patterns was determined to be 3.35 Å, corresponding to d110 of the cassiterite phase. Fig. 2(e) shows the selected area electron diffraction (SAED) pattern monitored over the entire area of Fig. 2(c)'s single particle. The SAED pattern, as is apparent, exhibited clear diffraction spots indexed to the tetragonal P42/mnm space group of the cassiterite phase, indicating that the prepared 10 mol% Sb-doped ATO nanoparticle was a single crystal without any impurity phases.
Fig. 3(a) shows the TEM image of the CdSe prepared in noncoordinating solvent ODE with the addition of TOPO.43 The as-prepared CdSe nanoparticles were monodispersed and of 6–7 nm size. The high resolution TEM image in Fig. 3(b) shows a CdSe QD with uniform fringe patterns over its entire area. The fringe spacing of 3.51 Å was determined to be d111 of the CdSe in the cubic F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group. Fig. 1(b) shows the XRD patterns of the pure CdSe, TiO2, ATO and ATO/TiO2/CdSe composites. The observed (111), (220), and (311) XRD peaks confirm that the prepared CdSe QD is in the pure cubic phase (JCPDS, No. 88-2346). The sol–gel derived TiO2, heat-treated at 250 °C, showed diffraction peaks at 25.31°, 37.79°, 48.05°, 55.07°, and 62.69°, corresponding to the (101), (004), (200), (211), and (204) peaks, respectively, of the anatase TiO2 (JCPDS, No. 71-1166). The XRD patterns of the ATO/TiO2/CdSe composites were a simple mixture of those of ATO, TiO2 and CdSe, without other impurity peaks, suggesting that no chemical reaction had occurred among the ATO, TiO2 and CdSe in the course of the heat-treatment at 250 °C.
3m space group. Fig. 1(b) shows the XRD patterns of the pure CdSe, TiO2, ATO and ATO/TiO2/CdSe composites. The observed (111), (220), and (311) XRD peaks confirm that the prepared CdSe QD is in the pure cubic phase (JCPDS, No. 88-2346). The sol–gel derived TiO2, heat-treated at 250 °C, showed diffraction peaks at 25.31°, 37.79°, 48.05°, 55.07°, and 62.69°, corresponding to the (101), (004), (200), (211), and (204) peaks, respectively, of the anatase TiO2 (JCPDS, No. 71-1166). The XRD patterns of the ATO/TiO2/CdSe composites were a simple mixture of those of ATO, TiO2 and CdSe, without other impurity peaks, suggesting that no chemical reaction had occurred among the ATO, TiO2 and CdSe in the course of the heat-treatment at 250 °C.

The TEM images in Fig. 4(a) show the 5/95 ATO/TiO2 composites prepared by the sol–gel method. The ATO particles of ∼50 nm size are fully covered with TiO2. Fig. 4(b) is a high-magnification image of the interface between TiO2 and ATO. The sharp interface without any chasm suggests that the TiO2 is tightly bound to the ATO particles.
 | ||
| Fig. 4 (a) TEM image of 5/95 ATO/TiO2, and (b) high-magnification image of interface between ATO and TiO2. | ||
A double heterojunction structure of ATO/TiO2/CdSe was formed by loading the CdSe QDs onto the surface of the 5/95 ATO/TiO2 composite. Scheme 2(a) is a schematic diagram of the preparation of the structure. Fig. 5(a) and 5(b) shows TEM images of the 1 wt% CdSe-loaded ATO/TiO2, clearly showing that the large ATO particles are located in the core and fully covered with less crystallized TiO2. The high magnification image (Fig. 5(c)) for the dotted rectangular part of c in Fig. 5(a), exhibits that the small CdSe quantum dots are scattered on the surface of the TiO2. Since the ATO particles were embedded in a large quantity of the sol–gel derived TiO2, the CdSe QDs and ATO particles were not in contact each other. They were spatially separated by the TiO2 layer.
 | ||
| Fig. 5 (a) TEM images of 1 wt% CdSe-loaded ATO/TiO2 composite, and (b) and (c) are the high resolution images magnifying the dotted rectangular areas of b and c, respectively, shown in (a). | ||
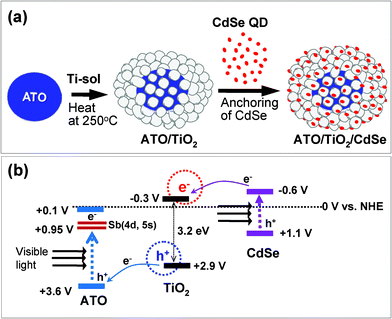 | ||
| Scheme 2 (a) Formation scheme for ATO/TiO2/CdSe double heterojunction system. (b) Energy band diagram indicating electron flows under visible-light irradiation. All of the potential levels versus NHE are indicated. | ||
EDX-mapping images were obtained for the 1 wt% CdSe-loaded ATO/TiO2 composite shown in the TEM image of Fig. 6(a). Fig. 6(b) shows an image of the Sn-mapping, clearly indicating that the dark particles embedded in the TiO2 structure (Fig. 6(a)) are aggregated ATO. The Ti-mapping image shown in Fig. 6(c) was very close to the original TEM image. Fig. 6(d) shows an image of the Cd-mapping. The overall mapping signal is very weak, but the image is close to that of Ti, suggesting that the CdSe QDs are uniformly distributed over the entire surface of TiO2 in ATO/TiO2.
 | ||
| Fig. 6 (a) TEM image of ATO/TiO2/CdSe (CdSe: 1 wt%) sample. EDX mapping images were obtained for the same area by monitoring the concentrations of (b) Sn, (c) Ti, and (d) Cd, respectively. | ||
Fig. 7 shows the UV-visible absorption spectra for the TiO2, CdSe, ATO, ATO/TiO2 and ATO/TiO2/CdSe composites. TiO2, due to its wide band gap, showed a low absorption in the visible region, whereas ATO revealed a profound absorption over the entire visible region. As a result, the ATO/TiO2 composites exhibited an appreciable absorption in the visible region. The characteristic absorption edge of CdSe QD, shown in the absorption spectra in Fig. 7, indicated that its band gap is 1.7 eV, which corresponds to the reported value of bulk CdSe.54–56 As a result, the ATO/TiO2/CdSe composite showed a significant absorption in the visible region, reflecting efficient utilization of visible light in the photocatalytic reactions.
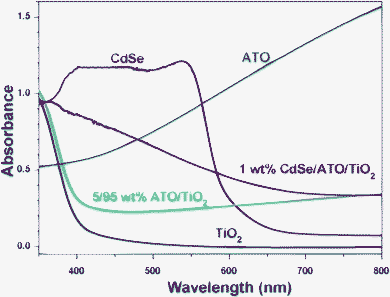 | ||
| Fig. 7 UV-visible absorption spectra of TiO2, CdSe, ATO, 5/95 ATO/TiO2, and ATO/TiO2/CdSe (CdSe: 1 wt%) powders, taken at diffuse reflectance mode. | ||
The photocatalytic activities of the CdSe/TiO2, ATO/TiO2 and ATO/TiO2/CdSe composites in decomposing gaseous IP were evaluated under visible-light irradiation (λ ≥ 420 nm). The photocatalytic reaction proceeded through initial oxidation of IP to acetone and then ultimate mineralization to CO2. Fig. 8(a) depicts the removal of the IP with several ATO/TiO2 composites of different compositions under visible-light irradiation. The photocatalytic activities of several ATO/TiO2 composites were remarkably higher than those of the end members such as the bare ATO nanoparticles or the blank TiO2 sample. Among them, the 5/95 ATO/TiO2 composite exhibited the highest catalytic activity. About 80% of IP was decomposed within 2 h of visible-light irradiation. When the ATO exceeded 5 wt%, however, the photocatalytic activity of the composites gradually decreased. The photocatalytic activity also was evaluated with respect to the evolution of CO2 under visible-light irradiation. As shown in Fig. 8(b), the 5/95 ATO/TiO2 composites showed the highest CO2-evolving efficiency. The amount of CO2 evolved in 2 h was 8.1 ppmv, whereas both the bare ATO and TiO2 showed negligible evolution.
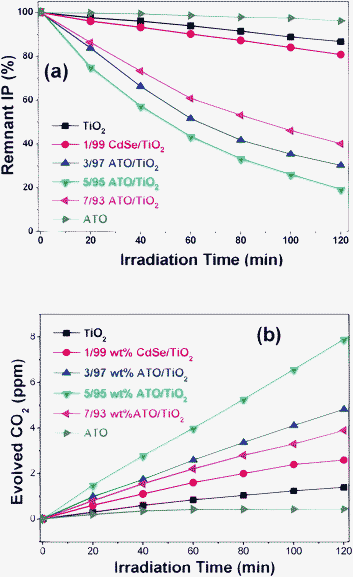 | ||
| Fig. 8 Photocatalytic decomposition of gaseous IP with ATO, TiO2, 1/99 CdSe/TiO2 and several ATO/TiO2 composites under visible-light irradiation. (a) Remnant IP percentage as function of irradiation time. (b) Amount of evolved CO2 (ppmv). | ||
For the purpose of comparison, several CdSe/TiO2 samples were prepared by loading different amounts of CdSe QD onto the surface of TiO2. Such are considered to be typical examples of the Type-A heterojunction structure, since the CB level of the CdSe is higher than that of TiO2, as shown in Scheme 1(a). It was found that 1/99 CdSe/TiO2 (wt% ratio) showed the optimum photocatalytic efficiency in decomposing IP and in evolving CO2. As shown in Fig. 8(a) and (b), its efficiency was appreciably higher than that of the bare CdSe or TiO2, but only ∼1/4 that of the 5/95 ATO/TiO2 composites in evolving CO2. The lower catalytic activity of CdSe/TiO2 would be caused by the fact that the holes, the most active component in the photocatalytic oxidation reaction, cannot be generated in the VB of TiO2. Under visible-light irradiation, the electrons of CdSe QDs are excited to its CB, and then transferred to the TiO2 CB. These electrons are useful to the reduction reaction, but will not be used efficiently in the oxidation reactions such as IP decomposition.
For further enhancement of visible-light photocatalytic activity, we prepared a double heterojunction structure by loading CdSe QDs onto the surface of 5/95 ATO/TiO2. Fig. 9(a) shows the photocatalytic activity of the CdSe-modified ATO/TiO2 composites in decomposing gaseous IP under visible light irradiation. The composites demonstrated remarkably enhanced catalytic activity, compared with the bare 5/95 ATO/TiO2. Among them, the 1.0 wt% CdSe loaded ATO/TiO2 composite exhibited the highest photocatalytic activity: about 97% of 2-propanal was decomposed in 2 h of irradiation. The photocatalytic activity also was evaluated according to the amount of CO2 evolved under visible-light irradiation, as shown in Fig. 9(b). The 1.0 wt% CdSe-loaded 5/95 ATO/TiO2 composites demonstrated the highest efficiency in evolving CO2: 23.2 ppmv after 2 h of irradiation, which is ∼3 times that of the bare 5/95 ATO/TiO2 composite. Its efficiency, indeed, was ∼13 times and ∼2 times that of Degussa P25 and the typical N-doped TiO2,33,37 respectively.
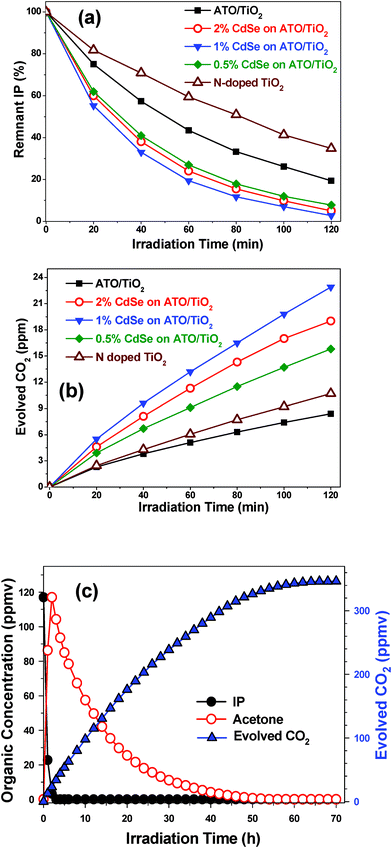 | ||
| Fig. 9 Photocatalytic decomposition of gaseous IP with ATO/TiO2, several ATO/TiO2/CdSe composites, and N-doped TiO2 under visible-light irradiation. (a) Remnant IP percentage as function of irradiation time. (b) Amount of evolved CO2 (ppmv). (c) Ultimate decomposition of IP with ATO/TiO2/CdSe (CdSe: 1.0 wt%) under long-term irradiation. | ||
Fig. 9(c) plots the trends of IP decomposition and CO2 evolution with 1.0 wt% CdSe loaded ATO/TiO2 composite under long-term visible-light irradiation. In 3 h, the IP, with an initial concentration of 117 ppm, was completely decomposed, while the acetone level was maximized at this time. Then, the acetone slowly disappeared, and in 70 h, the amount of evolved CO2 attained the maximum level. The measured CO2 level, 346 ppm, was close to the theoretical amount (351 ppm), suggesting that this photocatalytic system completely mineralized the IP without forming any intermediates.
Scheme 2b outlines the photocatalytic mechanism of the ATO/TiO2/CdSe composite system. Under visible-light irradiation, the electrons in the VB of ATO and CdSe are excited to their CBs. Then, the electrons in the VB of TiO2 can move to that of ATO, because the VB position of ATO lies below that of TiO2. At the same time, the photo-excited electrons in the CB of CdSe can move to that of TiO2, since the CB position of TiO2 lies below that of CdSe. As a result, both electrons and holes can be generated in the CB and VB of TiO2 under visible-light irradiation. In general, the photo-excited electron–hole pairs in bare TiO2 can be recombined quickly. In this double heterojunction structure, however, it is expected that the e−–h+ pairs generated in the TiO2 will have a relatively long lifetime, since the electrons and holes are space-charge-separated. In the ATO/TiO2/CdSe structure, ATO nanoparticles are embedded in the core, and CdSe QDs are loaded onto the surface of the TiO2 structure. Thus the electrons will be generated in the region neighboring the CdSe, whereas the holes will be formed near the ATO. Therefore, the space-charge-separated holes and electrons in TiO2 will have more opportunity to participate in photocatalytic reactions.
It was demonstrated in the present work that the double-heterojunction ATO/TiO2/CdSe composite, generating both electrons and holes on the surface of TiO2, demonstrates remarkably higher efficiency in decomposing IP and evolving CO2 than the ATO/TiO2 system which generates holes only. This strongly suggests that not only the holes in the VB of TiO2 but also the electrons in its CB play an important role in the complete decomposition of organic compounds. As indicated in eqn (3) and (4), HO2˙ can be formed from the electrons in the CB of TiO2. Therefore, both the HO˙ and HO2˙ will be the key components in inducing the decomposition reactions.
On the other hand, in the case of the ATO/TiO2/CdSe composite system under visible-light irradiation, the generated electrons in the CB of ATO and the holes in the VB of CdSe do not directly participate in oxidation reactions. In fact, it has been known that the holes in the VB of CdSe can be consumed by the adsorbed –OH group and/or induce photoanodic corrosion of CdSe,31 as shown in eqn (7). Then, what is the fate of the accumulated electrons in the CB of ATO? Considering that the CB level of ATO is considerably lower than the standard hydrogen potential, direct electron transfer to oxygen molecules, requiring −0.284 V (vs. NHE), will be difficult, as shown in eqn (8) . Thus we suppose that the electrons in the CB of ATO would be transported to oxygen species through the processes described in eqn (9)–(11). At present, we do not have concrete evidence for this. Presently mechanism studies are underway in order to fully understand the electron-transfer-passways in these novel heterojunction structures.
| nh+ + nCdSe → nSe + nCd2+ | (7) |
| O2 + e− → ˙O2−, E0 = −0.284 V (vs. NHE) | (8) |
| O2 + H+ + e− → HO2˙, E0 = −0.046 V | (9) |
| O2 + 2H+ + 2e− → H2O2, E0 = +0.682 V | (10) |
| O2 + 4H+ + 4e− → 2H2O, E0 = +1.23 V | (11) |
4. Conclusions
Deep blue-colored ATO (SbxSn1-xO2, x = 0.1) nanoparticles of ∼50 nm size were successfully prepared by co-precipitation and post-annealing at 1000 °C. The prepared ATO is a typical Type-B sensitizer, since it offers profound absorption in the visible range and its VB level is considerably lower than that of TiO2. By the hole-transfer mechanism, the 5/95 ATO/TiO2 composite exhibited notably high visible-light photocatalytic activity. The CO2 evolved in 2 h was 8.1 ppmv, whereas both the bare ATO and TiO2 showed negligible evolution. The double-heterojunction-structured 1% CdSe-loaded ATO/TiO2 composite demonstrated remarkably enhanced the photocatalytic activity in mineralization of IP. The amount of CO2 evolved was 23.2 ppmv after 2 h irradiation, which is ∼3 times and twice that of the bare ATO/TiO2 and the typical N-doped TiO2, respectively. The ultra-high visible-light photocatalytic activity of this novel double-heterojunction structure is due to the generation of both the electrons and holes generated in the CB and VB, respectively, of TiO2 under visible-light irradiation. Hence, it is suggested that not only the holes in the VB but also the electrons in the CB play an important role in the complete decomposition of organic compounds, and, further, that both the HO˙ and HO2˙ are key components in inducing such oxidation reactions.Acknowledgements
This work has been supported by the Korean Center for Artificial Photosynthesis (KCAP) funded by the Ministry of Education, Science, and Technology (NRF-2011-C1AAA001-2011-0030278), the Ministry of Environment (Project No. 022-061-026), and the National Research Foundation of Korea (Project No. 2011-0002995).References
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- E. S. Tang, J. H. Won, S. J. Hwang and J. H. Choy, Adv. Mater., 2006, 18, 3309 CrossRef.
- Z. Ding, G. Q. Lu and P. F. Greenfield, J. Phys. Chem. B, 2000, 104, 4815 CrossRef CAS.
- Y. Ou, J. Lin, S. Fang and D. Liao, Catal. Commun., 2007, 8, 936 CrossRef CAS.
- Y. Huang, Z. Zheng, Z. Ai, L. Zhang, X. Fan and Z. Zou, J. Phys. Chem. B, 2006, 110, 19323 CrossRef CAS.
- C. Burda, Y. Lou, X. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Nano Lett., 2003, 3, 1049 CrossRef CAS.
- V. Stengl, V. Houskova, S. Bakardjieva and N. Murafa, ACS Appl. Mater. Interfaces, 2010, 2, 575 CAS.
- S. Klosek and D. Raftery, J. Phys. Chem. B, 2001, 105, 2815 CrossRef CAS.
- S. Sakthivel and H. Kisch, ChemPhysChem, 2003, 4, 487 CrossRef CAS.
- K. Y. Song, M. K. Park, Y. T. Kwon, H. W. Lee, W. J. Chung and W. I. Lee, Chem. Mater., 2001, 13, 2349 CrossRef CAS.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505 CrossRef CAS.
- B. K. Vijayan, N. M. Dimitrijevic, J. Wu and K. A. Gray, J. Phys. Chem. C, 2010, 114, 21262 CAS.
- J. C. Xu, M. Lu, X. Y. Guo and H. L. Li, J. Mol. Catal. A: Chem., 2005, 226, 123 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669 CrossRef.
- P. Bouras, E. Stathatos and P. Lianos, Appl. Catal., B, 2007, 73, 51 CrossRef CAS.
- B. Peng, X. Meng, F. Tang, X. Ren, D. Chen and J. Ren, J. Phys. Chem. C, 2009, 113, 20240 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- F. Dong, H. Wang and Z. Wu, J. Phys. Chem. C, 2009, 113, 16717 CAS.
- T. Tachikawa, S. Tojo, K. Kawai, M. Endo, M. Fujitsuka, T. Ohno, K. Nishijima, Z. Miyamoto and T. Majima, J. Phys. Chem. B, 2004, 108, 19299 CrossRef CAS.
- W. Zhao, W. Ma, C. Chen, J. Zhao and Z. Shuai, J. Am. Chem. Soc., 2004, 126, 4782 CrossRef CAS.
- J. C. Yu, W. Ho, J. Yu, H. Yip, P. K. Wong and J. Zhao, Environ. Sci. Technol., 2005, 39, 1175 CrossRef CAS.
- T. Morikawa, Y. Irokawa and T. Ohwaki, Appl. Catal., A, 2006, 314, 123 CrossRef CAS.
- T. Morikawa, T. Ohwaki, K. Suzuki, S. Moribe and S. T. Kubota, Appl. Catal., B, 2008, 83, 56 CrossRef CAS.
- H. Irie, K. Kamiya, T. Shibanuma, S. Miura, D. A. Tryk, T. Yokoyama and K. Hashimoto, J. Phys. Chem. C, 2009, 113, 10761 CAS.
- D. Liu and P. V. Kamat, J. Electroanal. Chem., 1993, 347, 451 CrossRef CAS.
- L. Spanhel, H. Weller and A. Henglein, J. Am. Chem. Soc., 1987, 109, 6632 CrossRef CAS.
- X. Yu, Q. Wu, S. Jiang and Y. Guo, Mater. Charact., 2006, 57, 333 CrossRef CAS.
- J. C. Kim, J. K. Choi, Y. B. Lee, J. H. Hong, J. I. Lee, J. W. Yang, W. I. Lee and N. H. Hur, Chem. Commun., 2006, 5024 RSC.
- Y. Bessekhouad, N. Chaoui, M. Trzpit, N. Ghazzal, D. Robert and J. V. Weber, J. Photochem. Photobiol., A, 2006, 183, 218 CrossRef CAS.
- N. Serpone, P. Maruthamuthu, P. Pichat, E. Pelizzetti and H. Hidaka, J. Photochem. Photobiol., A, 1995, 85, 247 CrossRef CAS.
- S. Y. Chai, Y. J. Kim and W. I. Lee, J. Electroceram., 2006, 17, 909 CrossRef CAS.
- Y. J. Kim, B. Gao, S. Y. Han, M. H. Jung, A. K. Chakraborty, T. G. Ko, C. M. Lee and W. I. Lee, J. Phys. Chem. C, 2009, 113, 19179 CAS.
- S. B. Rawal, A. K. Chakraborty and W. I. Lee, Bull. Korean Chem. Soc., 2009, 30, 2613 CrossRef CAS.
- S. Y. Chai, Y. J. Kim, M. H. Jung, A. K. Chakraborty, D. W. Jung and W. I. Lee, J. Catal., 2009, 262, 144 CrossRef CAS.
- B. Gao, A. K. Chakraborty, J. M. Yang and W. I. Lee, Bull. Korean Chem. Soc., 2010, 31, 1941 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS.
- M. S. Wrighton, D. L. Morse, A. B. Ellis, D. S. Ginley and H. B. Abrahamson, J. Am. Chem. Soc., 1976, 98, 44 CrossRef CAS.
- Y. Xu and A. A. Schoonen, Am. Mineral., 2000, 85, 543 CAS.
- C. A. Vincent and D. G. C. Weston, J. Electrochem. Soc., 1972, 119, 518 CrossRef CAS.
- B. Orel, U. Lavrencis-Stangar, Z. Crnjak-Orel, P. Bukovec and M. Kosec, J. Non-Cryst. Solids, 1994, 167, 272 CrossRef CAS.
- F. Bai, Mater. Lett., 2006, 60, 3126 CrossRef CAS.
- H. J. Jeon, Mater. Lett., 2005, 59, 1801 CrossRef CAS.
- Q. Dai, D. Li, H. Chen, S. Kan, H. Li, S. Gao, Y. Hou, B. Liu and G. Zou, J. Phys. Chem. B, 2006, 110, 16509 Search PubMed.
- K. S. Leschkies, R. Divakar, J. Basu, E. E. Pommer, J. E. Boercker, C. B. Carter, U. R. Kortshagen, D. J. Norris and E. S. Aydil, Nano Lett., 2007, 7, 1793 CrossRef CAS.
- Y. T. Kwon, K. Y. Song, W. I. Lee, G. J. Choi and Y. R. Do, J. Catal., 2000, 191, 192 CrossRef CAS.
- Y. M. Cross and D. R. Pyke, J. Catal., 1979, 58, 61 CrossRef CAS.
- T. Nutz, U. Z. Felde and M. Haasea, J. Chem. Phys., 1999, 110, 12142 CrossRef CAS.
- V. Dusastre and D. E. Williams, J. Phys. Chem. B, 1998, 102, 6733 CrossRef.
- C. A. Vincent, J. Electrochem. Soc., 1972, 119, 515 CrossRef CAS.
- Z. Q. Li, Y. L. Yin, X. D. Liu, L. Y. Li, H. Liu and Q. G. Song, J. Appl. Phys., 2009, 106, 83701 CrossRef.
- N. Naghavi, C. Marcel, L. Dupont, J.-B. Leriche and J.-M. Tarascon, Solid State Ionics, 2003, 156, 463 CrossRef CAS.
- A. Klein, C. Körber, A. Wachqu, F. Säuberlich, Y. Gassenbauer, S. P. Harvey, D. E. Proffit and T. O. Mason, Materials, 2010, 3, 4892 CrossRef CAS.
- P. V. Kamat, Chem. Rev., 1993, 93, 207–300 CrossRef.
- C. Harris and P. V. Kamat, ACS Nano, 2009, 3, 682 CrossRef CAS.
- J. H. Bang and P. V. Kamat, ACS Nano, 2009, 3, 1467 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |