Platinum-catalyzed 1,3-acyloxy migration/[1,5]-hydride transfer/cycloaddition sequence: synthesis of ring-fused tetrahydroquinolines†
Xiao-Feng
Xia
a,
Xian-Rong
Song
a,
Ning
Wang
a,
Hai-Long
Wei
b,
Xue-Yuan
Liu
a and
Yong-Min
Liang
*a
aState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, 730000, China. E-mail: liangym@lzu.edu.cn; Fax: (+86) 931-8912582
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, P. R. China.
First published on 16th November 2011
Abstract
An efficient construction of multifunctionalized ring-fused tetrahydroquinolines viaplatinum-catalyzed C–H functionalization is presented. A sequence involving 1,3-OAc migration, [1,5]-hydride shift and then cyclization takes place to produce ring-fused tetrahydroquinolines. The reaction mechanism has been confirmed by a deuterium-labeling experiment.
Introduction
The development of methodologies for the direct functionalization of relatively less reactive C–H bonds, which provides a powerful tool for the synthesis of numerous complex molecules,1 has now become one of the most challenging projects in organic synthesis. Various hydrogen-exchange protocols have been proposed to explain the phenomenon, which are different from oxidative C–H bond functionalization by eliminating redox adjustment steps required to generate reactive functional groups for the desired C–C bond formation.1 Among these efficient methods, tandem [1,5]-hydride transfer/cyclization reaction is an attractive strategy, which has been recently reported by Sames and Gagosz as well as other research groups.2 In particular, tertiary anilines as hydride donors (tert-amine effect3) has been exploited by the Seidel group and others to synthesize tetrahydroquinolines. On the other hand, propargylic ester as the precursor of allene has been utilized to build lots of complex molecular scaffolds in platinum chemistry from infancy to mature.4 However, to the best of our knowledge, there are few reports about hydrofunctionalization of allene as a hydride accepter in a 1,5-hydride transfer/cyclization reaction (Scheme 1).5,12 Herein, we report a novel redox–neutral domino reaction using in situ formed allene to achieve multifunctionalized ring-fused tetrahydroquinoline.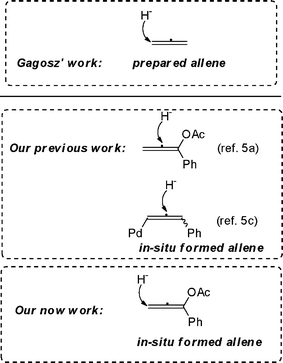 | ||
| Scheme 1 Hydrofunctionalization of allene. | ||
Our group is persistently interested in C–H functionalization at the α-position of tertiary amines to prepare various functionalized heterocyclic compounds.6 In conjunction with our recent investigations in the field of platinum-catalyzed hydride transfers, we were intrigued by the possibility of developing a new hydrofunctionalization of the in situ formed allenes. In contrast with a traditional hydrofunctionalization,4 such a process would first involve the transfer of a hydride onto the C(1)-carbon of the allene intermediate 2 activated by platinum formed viaplatinum-catalyzed 1,3-OAc migration of the propargylic ester 1,7 which is rarely reported (Scheme 2). The resulting vinyl-platinum species would then attack the iminium ion in a final electrophilic demetalation step to furnish the ring-fused tetrahydroquinoline compound which is the key structural element in many natural products, particularly alkaloids.8 We report herein a successful realization of this concept and the details of our discovery.
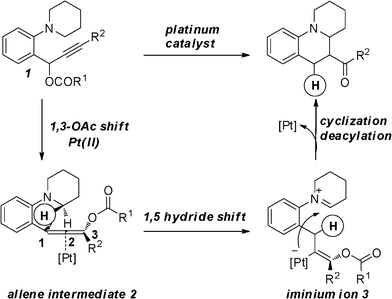 | ||
| Scheme 2 Proposed Lewis acid-catalyzed process. | ||
Results and discussion
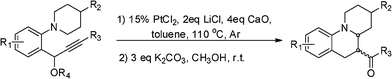
We started our initial study of reaction conditions using propargylic ester 1a as a model substrate (Table 1). When 1a was subjected to the reaction conditions used in our previous work (10 mol% of PtCl2, in toluene at 80 °C), an unstable cycloadduct was afforded. But due to the lack of diastereoselectivity and chromatographic separation, the initially formed cycloadduct was then subjected to deacylation with K2CO3 in MeOH. To our delight, an interesting ketone derivative 2a was isolated in 22% yield. The structure of compound 2a was elucidated on the basis of one- and two-dimensional NMR data (see supporting information†). Increasing the temperature and the loading of the catalyst led to a better yield (Table 1, Scheme 3). Among the platinum catalysts tried (Table 1, entries 4–7), PtCl2 (15 mol%) under Ar (1 atm) gave the best result (Table 1, entry 6). To further optimize the conditions, the influence of additives was investigated (Table 1, entries 8 and 9). Use of LiCl as an additive did enhance the catalytic activity of PtCl2,9 and the yield was increased to 65% (Table 1, entry 8). After this, we screened the amount of additive and found that 2eq LiCl gave a better result (Table 1, entry 8). The addition of M.S. or CaO could further improve the reaction yield (Table 1, entries 10 and 11). With 15 mol% of PtCl2 as the catalyst, 2 eq LiCl and 4 eq CaO as the additives, a better yield (78%) was obtained at 110 °C (Table 1, entry 12).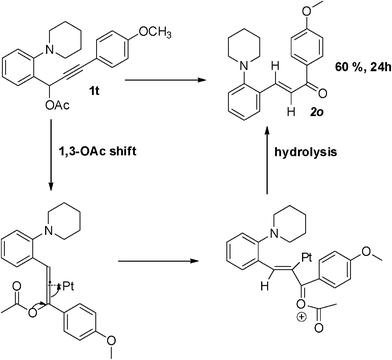 | ||
| Scheme 3 Plausible mechanism leading to the formation of 2o. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive | Solvent | T/°C | Yield (%)b |
| a Unless otherwise noted, all reactions were performed with 0.2 mmol of 1a in toluene (2.0 mL). b Isolated yields over two steps. | |||||
| 1 | PtCl2(10) | — | Toluene | 80 | 22 |
| 2 | PtCl2(15) | — | Toluene | 80 | 30 |
| 3 | PtCl2(10) | — | Toluene | 110 | 35 |
| 4 | PtCl2(15) | — | Toluene | 110 | 38 |
| 5 | PtCl2(15)/O2(1 atm) | — | Toluene | 110 | 46 |
| 6 | PtCl2(15)/Ar(1 atm) | — | Toluene | 110 | 56 |
| 7 | PtCl2(15)/CO(1atm) | — | Toluene | 110 | 40 |
| 8 | PtCl2(15)/Ar(1 atm) | LiCl (2 equiv) | Toluene | 110 | 65 |
| 9 | PtCl2(15)/Ar(1 atm) | LiCl (4 equiv) | Toluene | 110 | 57 |
| 10 | PtCl2(15)/Ar(1 atm) | LiCl (2 equiv)/4 Å MS (50 mg) | Toluene | 110 | 68 |
| 11 | PtCl2(15)/Ar(1 atm) | LiCl (2 equiv)/CaO (2 equiv) | Toluene | 110 | 70 |
| 12 | PtCl2(15)/Ar(1 atm) | LiCl (2 equiv)/ CaO (4 equiv) | Toluene | 110 | 78 |
| 13 | PtCl2(15)/Ar(1 atm) | LiCl (2 equiv)/CaO (6 equiv) | Toluene | 110 | 72 |
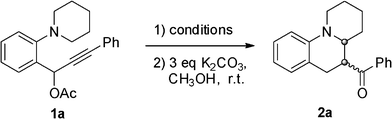
Various substrates were prepared to test the generality of this tandem sequence under the optimal conditions (Table 1, entry 12), and the results are summarized in Table 2. We extended the scope of this transformation to a wide range of electronically and structurally diverse propargylic esters. The 1,5-hydride shift proceeded smoothly for all of the tested propargylic esters to give the ring-fused tetrahydroquinolines in moderate to good yields with moderate dr values. Firstly, we screened the effect of migratory groupR4. To our delight, the acetate 1a, as an excellent substrate, afforded the highest yield, while benzoate 1b behaved better than 1c. The presence of an electron-withdrawing or -donating group in R3 was well tolerated. An electron-withdrawing substituent in R3 favored products formation (entries 7–10), while an electron-donating group slightly hindered the reaction (entries 4–6). This might be due to the fact that the electron-withdrawing substituent in R3 increases the cationic character of C1 in the allene intermediate (Scheme 2). To our delight, propargylic ester 1j was found to be equally a good candidate for the process and 78% of 2g was obtained with excellent dr value (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Substrate 1k bearing an electron-withdrawing bromine atom on the benzene ring (R1 = Br) gave the desired product 2h in 61% yield (entry 11). When both R1 and R3 were electron-withdrawing groups, a better result was achieved (entry 13). When R2 was methyl, the reactions also proceeded smoothly to give the corresponding products 2l and 2m in 60% and 50% yields, respectively. When propargylic ester 1r was subjected to the optimized conditions, the thienyl substituent in R3 was also tolerated and the product 2n was isolated in 63% yield. Unfortunately, sterically demanding ortho substituents could not participate in this reaction and acyclic tertiary aniline also failed to undergo this transformation (entry 19).
:1). Substrate 1k bearing an electron-withdrawing bromine atom on the benzene ring (R1 = Br) gave the desired product 2h in 61% yield (entry 11). When both R1 and R3 were electron-withdrawing groups, a better result was achieved (entry 13). When R2 was methyl, the reactions also proceeded smoothly to give the corresponding products 2l and 2m in 60% and 50% yields, respectively. When propargylic ester 1r was subjected to the optimized conditions, the thienyl substituent in R3 was also tolerated and the product 2n was isolated in 63% yield. Unfortunately, sterically demanding ortho substituents could not participate in this reaction and acyclic tertiary aniline also failed to undergo this transformation (entry 19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Substrates | Products | Time (h) | Yieldb (%)/d.r.c |
| a Unless otherwise noted, all reactions were performed with 0.2 mmol of propargylic esters in toluene (2.0 mL) at 110 °C. b Isolated yield after two steps. c Diastereomeric ratio is determined by 1H NMR spectroscopic analysis. d The yield is below 5%. | |||||
| 1 | 1a | R 1 = H, R2 = H, R3 = C6H5, R4 = Ac | 2a | 36 | 78 (2:1) |
| 2 | 1b | R 1 = H, R2 = H, R3 = C6H5, R4 = PhCO | 2a | 36 | 72 (6:1) |
| 3 | 1c | R 1 = H, R2 = H, R3 = C6H5, R4 = CH3CH2CO | 2a | 37 | 46 (4:1) |
| 4 | 1d | R 1 = H, R2 = H, R3 = p-MeC6H4, R4 = Ac | 2b | 35 | 50 (6:1) |
| 5 | 1e | R 1 = H, R2 = H, R3 = m-MeC6H4, R4 = Ac | 2c | 36 | 57 (13:1) |
| 6 | 1f | R 1 = H, R2 = H, R3 = 3,4-DiMeC6H3, R4 = Ac | 2d | 37 | 40 (1:1) |
| 7 | 1g | R 1 = H, R2 = H, R3 = p-ClC6H4, R4 = Ac | 2e | 38 | 63 (5:1) |
| 8 | 1h | R 1 = H, R2 = H, R3 = p-BrC6H4, R4 = Ac | 2f | 38 | 64 (8:1) |
| 9 | 1i | R 1 = H, R2 = H, R3 = p-BrC6H4, R4 = PhCO | 2f | 35 | 56 (5:1) |
| 10 | 1j | R 1 = H, R2 = H, R3 = p-CH3COC6H4 , R4 = Ac | 2g | 36 | 78 (>20:1) |
| 11 | 1k | R 1 = 5-Br, R2 = H, R3 = C6H5, R4 = Ac | 2h | 35 | 61 (6:1) |
| 12 | 1l | R 1 = 5-Br, R2 = H, R3 = C6H5, R4 = PhCO | 2h | 35 | 44 (4:1) |
| 13 | 1m | R 1 = 5-Br, R2 = H, R3 = p-BrC6H4, R4 = Ac | 2i | 35 | 73 (8:1) |
| 14 | 1n | R 1 = 5-Br, R2 = H, R3 = p-MeC6H4, R4 = Ac | 2j | 35 | 56 (4:1) |
| 15 | 1o | R 1 = 5-Br, R2 = H, R3 = 3,4-DiMeC6H3, R4 = Ac | 2k | 36 | 55 (3:1) |
| 16 | 1p | R 1 = H, R2 = Me, R3 = C6H5, R4 = Ac | 2l | 36 | 60 (3:1) |
| 17 | 1q | R 1 = H, R2 = Me, R3 = p-MeC6H4, R4 = Ac | 2m | 37 | 50 (6:1) |
| 18 | 1r | R 1 = H, R2 = H, R3 = 2-thienyl, R4 = Ac | 2n | 36 | 63 (4:1) |
| 19 | 1s |
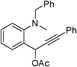
|
— | 30 | _d |
| 20 | 1t | R 1 = H, R2 = H, R3 = p-MeOC6H4, R4 = Ac |
20
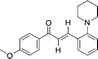
|
24 | 60 |
Surprisingly, a completely different transformation occurred in the case of propargylic ester 1t bearing a strong electron-donating 4–OMeC6H5 group in R3 and a α,β-unsaturated ketone 2o was formed (entry 20, Scheme 3), suggesting that in this case the rate of hydration of allene intermediate is faster than that of the 1,5-hydride shift. On the basis of these facts, it is confirmed that the presence of an electron-donating group in R3 is crucial to the success of the reaction.10
To explore the mechanism of these cycloisomerizations, a deuterium-labeling experiment was performed as shown in Scheme 4. Propargylic acetate D-1a containing CD2 produced cycloadduct D-2a in 50% yield. A mixture was isolated and the structures were assigned by 1H NMR analysis (see supporting information†). Analysis by 1H NMR indicated about 7 to 3 ratio of products with the compound D-2a-1 being major, in line with the previously reported deuterium isotope effect, indicating a faster rate of hydride than deuteride in the 1,5-H−/D− shift reaction.11 The deuterium atom in D-1a at the α-position of the nitrogen was transferred to the benzylic position of the D-2a, thus supporting a hydride transfer mechanism.
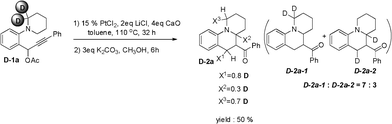 | ||
| Scheme 4 Deuterium-labeling experiment. | ||
Conclusions
In conclusion, we have developed a novel Lewis acid-catalyzed intramolecular redox domino reaction using 3-aryl-1-(2-(piperidin-1-yl)aryl)prop-2-ynyl esters to afford multifunctionalized ring-fused tetrahydroquinolines in moderate yields. Mechanistic studies revealed that the reaction might involve the transfer of a hydride adjacent to the nitrogen atom onto the in situ formed allene. This fascinating strategy allows the efficient conversion of tertiary sp3 C–H bonds into C–C bonds by the nucleophilic addition of a vinylplatinum species onto a zwitterionic intermediate.Experimental
General experimental
Column chromatography was carried out on silica gel. Unless noted 1H NMR spectra were recorded on 400 MHz or 300 MHz in CDCl3,13C NMR spectra were recorded on 100 MHz or 75 MHz in CDCl3 using TMS as internal standard. IR spectra were recorded on an FT-IR spectrometer and only major peaks are reported in cm−1. Melting points were determined on a microscopic apparatus and were uncorrected. All products were further characterized by HRMS (high resolution mass spectra); copies of their 1H NMR and 13C NMR spectra are provided.† Commercially available reagents and solvents were used without further purification.Specific experimental
2a was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.98–8.00 (m, 2 H), 7.59 (t, J = 7.2 Hz, 1 H), 7.46–7.49 (m, 2 H), 7.12 (t, J = 7.6 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.65 (t, J = 7.2 Hz, 1 H), 3.99–4.02 (m, 1 H), 3.71–3.78 (m, 1 H), 3.35–3.40 (m, 1 H), 2.96–3.03 (m, 1 H), 2.80–2.86 (m, 2 H), 1.71–1.77 (m, 3 H), 1.36–1.65 (m, 3 H); 13C NMR (100 MHz, CDCl3): δ 202.2, 146.1, 136.9, 133.3, 128.7, 128.6, 128.5, 128.3, 127.5, 123.1, 117.2, 112.9, 58.9, 48.5, 46.9, 32.4, 31.6, 25.4, 24.3; IR (neat, cm−1) 3399, 2920, 2852, 1677, 1585, 1450, 1057, 754, 705, 590; HRMS (ESI) m/z: calcd for C20H21NO: M + H = 292.1696; found: 292.1694. minor diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 7.2 Hz, 2 H), 7.52–7.56 (m, 1 H), 7.43 (t, J = 8.0 Hz, 2 H), 7.06–7.12 (m, 2 H), 6.65 (t, J = 7.2 Hz, 1 H), 6.49 (d, J = 7.6 Hz, 1 H), 3.84–3.89 (m, 1 H), 3.63–3.66 (m, 1 H), 3.38 (dd, J = 9.6, 8.0 Hz, 1 H), 3.27–3.32 (m, 1 H), 3.03 (dd, J = 11.6, 6.0 Hz, 1 H), 2.56–2.63 (m, 1 H), 1.84–1.86 (m, 1 H), 1.66–1.72 (m, 2 H), 1.34–1.59 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 199.2, 151.5, 137.0, 133.1, 133.0, 128.5, 128.0, 127.7, 124.5, 118.1, 106.6, 67.5, 45.8, 39.1, 38.5, 26.3, 24.5, 24.4;
2b was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.90 (d, J = 8.0 Hz, 2 H), 7.25–7.29 (m, 2 H), 7.12 (t, J = 7.6 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.65 (t, J = 7.6 Hz, 1 H), 3.99–4.02 (m, 1 H), 3.69–3.75 (m, 1 H), 3.32–3.38 (m, 1 H), 2.96–3.02 (m, 1 H), 2.78–2.86 (m, 2 H), 2.42 (s, 3 H), 1.71–1.77 (m, 3 H), 1.56–1.62 (m, 1 H), 1.26–1.47 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 201.8, 146.1, 144.2, 134.6, 129.5, 128.7, 128.5, 127.5, 123.2, 117.2, 112.9, 58.9, 48.5, 46.9, 32.6, 31.6, 25.5, 24.3, 21.6; IR (neat, cm−1) 3434, 2925, 2852, 1673, 1604, 1494, 1451, 1377, 1255, 1225, 1177, 1113, 1061, 747, 574; HRMS (ESI) m/z: calcd for C21H23NO: M + H = 306.1852; found: 306.1848.
2c was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.78–7.80 (m, 2 H), 7.34–7.41 (m, 2 H), 7.12 (t, J = 7.6 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.65 (t, J = 7.2 Hz, 1 H), 3.99–4.02 (m, 1 H), 3.71–3.77 (m, 1 H), 3.33–3.39 (m, 1 H), 2.95–3.02 (m, 1 H), 2.79–2.86 (m, 2 H), 2.41 (s, 3 H), 1.71–1.77 (m, 3 H), 1.55–1.65 (m, 1 H), 1.23–1.49 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 202.5, 146.1, 138.6, 137.1, 134.1, 128.9, 128.7, 128.6, 127.5, 125.6, 123.2, 117.2, 112.9, 58.9, 48.5, 47.1, 32.6, 31.6, 25.5, 24.3, 21.3; IR (neat, cm−1) 3336, 2929, 2851, 1675, 1601, 1494, 1451, 1370, 1253, 1161, 1050, 744, 550; HRMS (ESI) m/z: calcd for C21H23NO: M + H = 306.1852; found: 306.1850.
2d was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.73–7.77 (m, 2 H), 7.14–7.26 (m, 1 H), 7.10–7.14 (m, 1 H), 6.94 (d, J = 7.6 Hz, 1 H), 6.87 (d, J = 8.4 Hz, 1 H), 6.63–6.67 (m, 1 H), 3.99–4.03 (m, 1 H), 3.69–3.76 (m, 1 H), 3.32–3.37 (m, 1 H), 2.96–3.02 (m, 1 H), 2.77–2.86 (m, 2 H), 2.33 (s, 3 H), 2.32 (s, 3 H), 1.71–1.77 (m, 3 H), 1.56–1.62 (m, 1 H), 1.24–1.47 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 202.1, 146.1, 143.0, 137.2, 135.0, 130.0, 129.5, 128.8, 127.4, 126.1, 123.3, 117.1, 112.9, 59.0, 48.5, 46.8, 32.6, 31.6, 25.6, 24.3, 20.0, 19.8; IR (neat, cm−1) 3400, 2923, 2851, 1671, 1602, 1494, 1451, 1378, 1253, 1115, 1054, 793, 749, 708; HRMS (ESI) m/z: calcd for C22H25NO: M + H = 320.2009; found: 320.2006.
2e was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.91–7.94 (m, 2 H), 7.44–7.46 (m, 2 H), 7.12 (t, J = 7.6 Hz, 1 H), 6.93 (d, J = 7.6 Hz, 1 H), 6.85 (d, J = 8.4 Hz, 1 H), 6.66 (t, J = 7.2 Hz, 1 H), 3.98–4.01 (m, 1 H), 3.65–3.71 (m, 1 H), 3.32–3.38 (m, 1 H), 2.95–3.01 (m, 1 H), 2.78–2.86 (m, 2 H), 1.55–1.76 (m, 4 H), 1.38–1.49 (m, 1 H), 1.19–1.29 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 200.9, 146.0, 139.9, 135.3, 129.7, 129.1, 128.7, 127.6, 122.9, 117.3, 112.9, 58.8, 48.5, 47.1, 32.3, 31.6, 25.4, 24.3; IR (neat, cm−1) 3436, 2929, 2851, 1677, 1590, 1493, 1451, 1253, 1092, 1008, 839, 747, 534; HRMS (ESI) m/z: calcd for C20H20ClNO: M + H = 326.1306; found: 326.1301.
2f was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 8.4 Hz, 2 H), 7.62 (d, J = 8.4 Hz, 2 H), 7.12 (t, J = 7.6 Hz, 1 H), 6.93 (d, J = 7.6 Hz, 1 H), 6.85 (d, J = 8.4 Hz, 1 H), 6.65 (t, J = 7.2 Hz, 1 H), 3.98–4.01 (m. 1 H), 3.64–3.69 (m, 1 H), 3.33–3.38 (m, 1 H), 2.95–3.01 (m, 1 H), 2.78–2.86 (m, 2 H), 1.56–1.76 (m, 4 H), 1.20–1.49 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 201.1, 146.0, 135.7, 132.1, 129.8, 128.7, 128.6, 127.6, 122.9, 117.3, 112.9, 58.8, 48.5, 47.1, 32.3, 31.6, 25.4, 24.3; IR (neat, cm−1) 3403, 2923, 2853, 1764, 1681, 1452, 1381, 1243, 1061, 913, 746; HRMS (ESI) m/z: calcd for C20H20NOBr: M + H = 370.0801; found: 370.0806.
2g was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 8.05 (m, 4 H), 7.13 (t, J = 7.6 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.66 (t, J = 7.2 Hz, 1 H), 3.99–4.02 (m, 1 H), 3.72–3.78 (m, 1 H), 3.35–3.41 (m, 1 H), 2.96–3.03 (m, 1 H), 2.80–2.86 (m, 2 H), 2.65 (s, 3 H), 1.73–1.79 (m, 3 H), 1.28–1.69 (m, 3 H); 13C NMR (100 MHz, CDCl3): δ 201.7, 197.4, 146.0, 140.2, 140.1, 128.7, 128.6, 128.5, 127.6, 122.8, 117.3, 113.0, 58.8, 48.5, 47.6, 32.2, 31.6, 26.9, 25.3, 24.3; IR (neat, cm−1) 2927, 2852, 1683, 1601, 1495, 1259, 910, 748; HRMS (ESI) m/z: calcd for C22H23NO2: M + H = 334.1802; found: 334.1812.
2h was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.96–7.98 (m, 2 H), 7.59 (t, J = 7.2 Hz, 1 H), 7.47–7.50 (m, 2 H), 7.17–7.19 (m, 1 H), 7.03 (d, J = 2.0 Hz, 1 H), 6.69 (d, J = 8.8 Hz, 1H); 3.89–3.93 (m, 1 H), 3.67–3.72 (m, 1 H), 3.34–3.39 (m, 1 H), 2.91–2.97 (m, 1 H), 2.76–2.86 (m, 2 H), 1.70–1.79 (m, 3 H), 1.54–1.63 (m, 1 H), 1.39–1.49 (m, 1 H), 1.20–1.30 (m, 1 H); 13C NMR (100 MHz, CDCl3): δ 201.6, 145.1, 136.8, 133.4, 130.9, 130.1, 128.8, 128.3, 125.3, 114.5, 108.8, 58.7, 48.4, 46.8, 31.8, 31.5, 25.2, 24.2; IR (neat, cm−1) 3436, 2927, 2851, 1676, 1592, 1489, 1445, 1370, 1254, 1219, 793, 705, 546; HRMS (ESI) m/z: calcd for C20H20NOBr: M + H = 370.0801; found: 370.0795.
2i was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 8.4 Hz, 2 H), 7.63 (d, J = 8.8 Hz, 2 H), 7.17–7.19 (m, 1 H), 7.03 (d, J = 2.0 Hz, 1 H), 6.69 (d, J = 8.8 Hz, 1 H), 3.89–3.93 (m, 1 H), 3.60–3.66 (m, 1 H), 3.31–3.36 (m, 1 H), 2.90–2.97 (m, 1 H), 2.74–2.85 (m, 2 H), 1.66–1.79 (m, 3 H), 1.28–1.63 (m, 3 H); 13C NMR (100 MHz, CDCl3): δ 200.6, 145.0, 135.4, 132.2, 130.9, 130.1, 129.8, 128.8, 125.0, 114.5, 108.9, 58.7, 48.4, 46.8, 31.7, 31.5, 25.1, 24.2; IR (neat, cm−1) 3433, 2923, 2851, 1764, 1676, 1584, 1488, 1387, 1251, 1066, 1009, 746; HRMS (ESI) m/z: calcd for C20H19NOBr2: M + H = 447.9906; found: 447.9914.
2j was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 8.4 Hz, 2 H), 7.29 (d, J = 8.0 Hz, 2 H), 7.18 (dd, J = 6.8, 2.0 Hz, 1 H), 7.03 (d, J = 2.0 Hz, 1 H), 6.70 (d, J = 8.8 Hz, 1 H), 3.90–3.93 (m, 1 H), 3.64–3.70 (m, 1 H), 3.32–3.38 (m, 1 H), 2.91–2.98 (m, 1 H), 2.74–2.86 (m, 2 H), 2.43 (s, 3 H), 1.69–1.79 (m, 3 H), 1.26–1.59 (m, 3 H); 13C NMR (100 MHz, CDCl3): δ 201.2, 145.2, 144.4, 134.4, 130.9, 130.1, 129.5, 128.5, 125.4, 114.5, 108.8, 58.8, 48.5, 46.7, 31.9, 31.5, 25.2, 24.2, 21.6; IR (neat, cm−1) 3400, 2922, 2852, 2332, 1764, 1674, 1489, 1380, 1248, 1058, 913, 771, 747, 669; HRMS (ESI) m/z: calcd for C21H22NOBr: M + H = 384.0958; found: 384.0962.
2k was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.67–7.76 (m, 2 H), 7.10–7.25 (m, 3 H), 6.28–6.34 (m, 1 H), 3.81–3.87 (m, 1 H), 3.59–3.56 (m, 1 H), 3.23–3.37 (m, 2 H), 2.95–3.01 (m, 1 H), 2.57–2.64 (m, 1 H), 2.32 (s, 3 H), 2.31 (s, 3 H), 1.83–1.85 (m, 1 H), 1.31–1.69 (m, 5 H); 13C NMR (100 MHz, CDCl3): δ 198.5, 150.5, 142.7, 136.9, 135.6, 134.7, 130.2, 129.9, 129.8, 129.2, 127.5, 127.1, 125.8, 125.7, 109.5, 107.8, 67.5, 45.7, 38.9, 38.1, 26.0, 24.2, 24.1, 20.0, 19.8; IR (neat, cm−1) 2930, 2852, 1679, 1602, 1477, 1448, 1382, 1249, 1122, 803; HRMS (ESI) m/z: calcd for C22H24BrNO: M + H = 398.1114; found: 398.1111.
2l was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.99–8.01 (m, 2 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.49 (t, J = 7.6 Hz, 2 H), 7.12 (t, J = 7.8 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H),6.65 (t, J = 7.2 Hz, 1 H), 3.99–4.04 (m, 1 H), 3.70–3.76 (m, 1 H), 3.38–3.43 (m, 1 H), 2.94–3.00 (m, 1 H), 2.81–2.88 (m, 2 H), 1.70–1.76 (m, 2 H), 1.21–1.64 (m, 3 H), 0.87 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ 202.2, 145.9, 136.9, 133.3, 128.8, 128.6, 128.4, 127.5, 123.1, 117.2, 112.9, 58.3, 48.0, 47.1, 39.9, 33.8, 32.6, 30.9, 21.9; IR (neat, cm−1) 3440, 2923, 2849, 1677, 1600, 1495, 1452, 1373, 1239, 1210, 750, 702, 665; HRMS (ESI) m/z: calcd for C21H23NO: M + H = 306.1852; found: 306.1855.
2m was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.89–7.92 (m, 2 H), 7.25–7.30 (m, 2 H), 7.12 (t, J = 7.8 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.65 (t, J = 7.2 Hz, 1 H), 3.99–4.04 (m, 1 H), 3.68–3.74 (m, 1 H), 3.36–3.41 (m, 1 H), 2.94–3.01 (m, 1 H), 2.79–2.88 (m, 2 H), 2.43 (s, 3 H), 1.69–1.77 (m, 2 H), 1.53–1.63 (m, 2 H), 1.24–1.28 (m, 1 H), 0.87 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ 201.8, 145.9, 144.3, 134.5, 129.5, 128.7, 128.5, 127.5, 123.2, 117.2, 112.9, 58.4, 48.1, 46.9, 39.9, 33.9, 32.7, 30.9, 21.9, 21.6; IR (neat, cm−1) 3399, 2019, 2850, 1672, 1603, 1494, 1453, 1374, 1241, 1045, 748, 610; HRMS (ESI) m/z: calcd for C22H25NO: M + H = 320.2009; found: 320.2001.
2n was obtained according to the above method as an oil. major diastereomer: 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 3.6 Hz, 1 H), 7.68 (d, J = 4.8 Hz, 1 H), 7.10–7.17 (m, 2 H), 6.96 (d, J = 7.2 Hz, 1 H), 6.87 (d, J = 8.4 Hz, 1 H), 6.67 (t, J = 7.2 Hz, 1 H), 3.99–4.02 (m, 1 H), 3.50–3.56 (m, 1 H), 3.28–3.34 (m, 1 H) 3.05–3.12 (m, 1 H), 2.78–2.87 (m, 2 H), 1.76–1.78 (m, 3 H), 1.57–1.66 (m, 1 H), 1.28–1.47 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 194.9, 146.1, 144.9, 134.5, 132.2, 128.6, 128.4, 127.5, 123.0, 117.3, 113.1, 58.8, 49.1, 48.5, 32.6, 31.6, 25.5, 24.1; IR (neat, cm−1) 3399, 2925, 2851, 1653, 1494, 1413, 1253, 1057, 789, 744, 661; HRMS (ESI) m/z: calcd for C18H19NOS: M + H = 298.1260; found: 298.1254.
2o 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 15.6 Hz, 1 H), 8.05 (d, J = 8.8 Hz, 2 H), 7.66 (d, J = 8.4 Hz, 1 H), 7.52 (d, J = 16.0 Hz, 1 H), 7.32–7.37 (m, 1 H), 7.05 (d, J = 7.6 Hz, 2 H), 6.98 (d, J = 8.8 Hz, 2 H), 3.89 (s, 3 H), 2.91–2.94 (m, 4 H), 1.73–1.78 (m, 4 H), 1.57–1.60 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ 189.4, 163.2, 154.4, 142.0, 131.4, 130.9, 130.8, 130.7, 129.2, 127.9, 122.3, 121.4, 119.0, 113.7, 113.6, 55.4, 54.4, 26.4, 24.2; IR (neat, cm−1): 2928, 1655, 1599, 1449, 1380, 1332, 1257, 1219, 1167, 1021, 835, 757, 575; HRMS (ESI) m/z: calcd for C21H23NO2: M + H = 322.1802; found: 322.1808.
D–2a1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 7.2 Hz, 2 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.49 (t, J = 8.0 Hz, 2 H), 7.13 (t, J = 7.6 Hz, 1 H), 6.94 (d, J = 7.2 Hz, 1 H), 6.86 (d, J = 8.0 Hz, 1 H), 6.65 (t, J = 7.2 Hz, 1 H), 3.99–4.02 (m, 0.3 H), 3.71–3.78 (m, 1 H), 3.35–3.40 (m, 0.7 H), 2.96–3.03 (m, 1 H), 2.80–2.88 (m, 1.2 H), 1.38–1.79 (m, 6 H); 13C NMR (100 MHz, CDCl3): δ 202.2, 146.1, 137.1, 133.3, 128.8, 128.7, 128.4, 127.5, 123.1, 117.2, 112.9, 58.9, 47.1, 32.5, 31.6, 25.3, 24.3; HRMS (ESI) m/z: calcd for C20H19NOD2: M + H = 294.1821; found: 294.1825.
Acknowledgements
We thank the National Science Foundation (NSF 21072080), and the Fundamental Research Funds for the Central Universities (lzujbky-2011-151 and lzujbky-2010-k09) for financial support. We acknowledge National Basic Research Program of China (973 Program) 2010CB833203 and “111” program of MOE.References
- (a) Recent reviews: F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 Search PubMed; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS; (c) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069 RSC; (d) L.-C. Campeau and K. Fagnou, Chem. Rev., 2007, 107, 174 CrossRef CAS; (e) R. Giri, B.-F. Shi, K. E. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (f) R. Jazzar, J. Hitch, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS; (g) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS.
- (a) For selected recent references, see: S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180 Search PubMed; (b) S. J. Pastine and D. Sames, Org. Lett., 2005, 7, 5429 CrossRef CAS; (c) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2006, 45, 1683 CrossRef CAS; (d) J. Barluenga, M. Fananás-Mastral, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2008, 47, 6594 CrossRef CAS; (e) S. Murarka, C. Zhang, M. D. Konieczynska and D. Seidel, Org. Lett., 2009, 11, 129 CrossRef CAS; (f) J. C. Ruble, A. R. Hurd, T. A. Johnson, D. A. Sherry, M. R. Barbachyn, P. L. Toogood, G. L. Bundy, D. R. Graber and G. M. Kamilar, J. Am. Chem. Soc., 2009, 131, 3991 CrossRef CAS; (g) K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2009, 131, 402 CrossRef CAS; (h) D. Shikanai, H. Murase, T. Hata and H. Urabe, J. Am. Chem. Soc., 2009, 131, 3166 CrossRef CAS; (i) S. Murarka, I. Deb, C. Zhang and D. Seidel, J. Am. Chem. Soc., 2009, 131, 13226 CrossRef CAS; (j) P. A. Vadola and D. Sames, J. Am. Chem. Soc., 2009, 131, 16525 CrossRef CAS; (k) I. D. Jurberg, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2010, 132, 3543 CrossRef CAS; (l) G. Zhou and J. Zhang, Chem. Commun., 2010, 46, 6593 RSC; (m) Y. K. Kang, S. M. Kim and D. Y. Kim, J. Am. Chem. Soc., 2010, 132, 11847 CrossRef CAS.
- (a) W. D. Verboom, N. Reinhoudt, R. Visser and S. Harkema, J. Org. Chem., 1984, 49, 269 CrossRef CAS; (b) W. H. N. Nijhuis, W. Verboom and D. N. Reinhoudt, J. Am. Chem. Soc., 1987, 109, 3136 CrossRef CAS; (c) W. H. N. Nijhuis, W. Verboom, A. A. El-Fadl, S. Harkema and D. N. Reinhoudt, J. Org. Chem., 1989, 54, 199 CrossRef CAS; (d) W. H. N. Nijhuis, W. Verboom, A. A. El-Fadl, G. J. Van Humme1 and D. N. Reinhoudt, J. Org. Chem., 1989, 54, 209 CrossRef CAS; (e) K. Mori, Y. Ohshima, K. Ehara and T. Akiyama, Chem. Lett., 2009, 38, 524 CrossRef CAS.
- (a) For selected reviews of propargylic esters in Pt catalysis, see: A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 Search PubMed; (b) J. Marco-Contelles and E. Soriano, Chem.–Eur. J., 2007, 13, 1350 CrossRef CAS; (c) L. Zhang, J.-W. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271 CrossRef CAS; (d) E. Soriano and J. Marco-Contelles, Acc. Chem. Res., 2009, 42, 1026 CrossRef CAS.
- (a) Allene as hydride accepter in 1,5-hydride transfer/cyclization reaction using Lewis acid as catalyst, see: X.-Z. Shu, K.-G. Ji, S.-C. Zhao, Z.-J. Zheng, J. Chen, L. Lu, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2008, 14, 10556 Search PubMed; (b) B. Bolte and F. Gagosz, J. Am. Chem. Soc., 2011, 133, 7696 Search PubMed; (c) S.-C. Zhao, X.-Z. Shu, K.-Go. Ji, A.-X. Zhou, T. He, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2011, 76, 1941 Search PubMed.
- (a) X.-Z. Shu, X.-F. Xia, Y.-F. Yang, K.-G. Ji, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2009, 74, 7464 CrossRef CAS; (b) X.-F. Xia, X.-Z. Shu, K.-G. Ji, Y.-F. Yang, S. Ali, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2010, 75, 2893 CrossRef CAS; (c) X.-F. Xia, X.-Z. Shu, K.-G. Ji, S. Ali, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2011, 76, 342 Search PubMed.
- (a) Based on our previous work, it is considered that platinum-Π-allene species could play a cationic role mainly at the C(1)-and C(3)-carbons, which can induce hydride shift smoothly, driven by the generation of a stable carbocation adjacent to the heteroatom. For metal-allene bonding having the dipolar character, see selected examples: H. Kusama, M. Ebisawa, H. Funami and N. Iwasawa, J. Am. Chem. Soc., 2009, 131, 16352 Search PubMed; (b) J. H. Lee and F. D. Toste, Angew. Chem., Int. Ed., 2007, 46, 912 CrossRef CAS; (c) G. Lemiere, V. Gandon, K. Cariou, A. Hours, T. Fukuyama, A.-L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2009, 131, 2993 CrossRef CAS; (d) T.-M. Teng and R.-S. Liu, J. Am. Chem. Soc., 2010, 132, 9298 Search PubMed.
- D. H. Barton, K. Nakanishi, O. Meth-Cohn, Comprehensive Natural Products Chemistry, Elsevier, Oxford, 1999, vols. 1–9 Search PubMed.
- LiCl likely serves to enhance the π-Lewis acidity of Li[PtCl3], see: K. Miura, G. Inoue, H. Sasagawa, H. Kinoshita, J. Ichikawa and A. Hosomi, Org. Lett., 2009, 11, 5066 Search PubMed.
- (a) For selected recent references, see: M. Yu, G. Zhang and L. Zhang, Org. Lett., 2007, 9, 2147 Search PubMed; (b) L. Ye and L. Zhang, Org. Lett., 2009, 11, 3646 CrossRef CAS; (c) M. Yu, G. Zhang and L. Zhang, Tetrahedron, 2009, 65, 1846 CrossRef CAS; (d) D.-W. Wang, X.-H. Ye and X.-D. Shi, Org. Lett., 2010, 12, 2088 CrossRef CAS.
- In reference 2f, the ratio of product derived from a hydrogen migration to product derived from deutero migration was∼3:1.
- One of the reviewers of this manuscript is of the view that this work is an extension of our previous work.5c However, we have excluded this because when we treated 1a with a palladium catalyst, we observed no reaction. The present reaction is completely different from that reported previously in terms of substrate structure, reaction conditions, catalyst and most importantly by mechanism.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00789k |
| This journal is © The Royal Society of Chemistry 2012 |