Synthesis and X-ray diffraction structures of novel half-sandwich Os(II)-and Ru(II)-hydroxamate complexes†
Attila J.
Godó
a,
Attila Cs.
Bényei
b,
Brian
Duff
cd,
Denise A.
Egan
cd and
Péter
Buglyó
*a
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, H-4010 Debrecen, Hungary. E-mail: buglyo@science.unideb.hu
bLaboratory for X-ray Diffraction, Institute of Chemistry, Univesity of Debrecen, H-4010 Debrecen, Hungary
cCentre for Pharmaceutical Research and Development, Institute of Technology Tallaght, Dublin 24, Ireland.
dDepartment of Science, Institute of Technology Tallaght, Dublin 24, Ireland.
First published on 20th December 2011
Abstract
Novel water soluble half-sandwich complexes of the general formulae [M(η6-p-cym)(ha)]2(CF3SO3)2, [M(η6-p-cym)(ha)Cl] or [M(η6-p-cym)(ha)(py)]X (M = Os, Ru; ha = hydroxamate; py = pyridine; X = Cl− or CF3SO3−), incorporating metal-containing entities and hydroxamates both with potential anti-proliferative features, were prepared and characterized by elemental analysis, spectroscopy (NMR, IR) and ESI mass spectrometry. The X-ray crystal structure of [Ru(η6-p-cym)(μ-meaha)]2(CF3SO3)2 (5), [Os(η6-p-cym)(meaha)Cl] (6), [Ru(η6-p-cym)(phebha)Cl], (9), [Ru(η6-p-cym)(bha)(py)](CF3SO3) (12) and [Ru(η6-p-cym)(phebha)(py)](CF3SO3) (14), 6 is the first published structure of an organometallic Os(II)-hydroxamate reported. The effect of size differences of the metal ions, the steric demand of the RC and RN substituents at the hydroxamate group and the type of the monodentate ligand co-present in the stoichiometry, along with the binding architecture of the half-sandwich metal(II) hydroxamate complexes are discussed. A novel dinuclear, dihydroxo bridged complex [Os(η6-p-cym)(py)(μ-OH)]2(CF3SO3)2 (16) is prepared and characterized by X-ray crystallography. Unexpected formation of a dinuclear oxo bridged OsII/OsVI complex [{Os(η6-p-cym)(meaha)}(μ-O){Os(O)(meaha)2}]Cl (17) occurs, and the crystal and molecular structure has been determined by X-ray method. Complexes 1, 5–8, 10 and 14 were tested for their in vitro cytotoxicity, using human-derived ovarian cancer cell lines (A2780 and A2780 cisR), and showed no anti-proliferative effect in the concentration range (0–200 μM) studied.
1. Introduction
Hydroxamic acids, R1CON(R2)OH, are an important class of biomolecules, capable of forming stable five-membered (O,O) chelates with a wide range of metal ions. This strong interaction may result in an essential role for these ligands in terms of uptake and transport of different metal ions, e.g. Fe3+, mainly in microorganisms, or in the effective and selective inhibition of various metalloenzymes.1 Based on the inhibition of histone deacetylases, a monohydroxamic acid, suberoilanilide hydroxamic acid (sahaH), is currently undergoing clinical use as a treatment for cutaneous T-cell lymphoma.2,3Half-sandwich Ru(II) complexes with promising anti-proliferative properties have also been the subject of intensive research in recent decades. Among others, the effects of the size and hydrophobicity of the η6-arene, the type of the coordinated (N,N) (N,O) or (O,O) chelating ligands, the rate of aquation of the monodentate ligand at the sixth coordination site, and the acidity of the water molecule after aquation have all been studied on the in vitro cytotoxicity of the complexes.4–7
We have hypothesized that a combination of the two entities, namely hydroxamate and [RuII(η6-arene)], into one molecule may result in the production of molecules with beneficial properties.8,9 Indeed, preliminary results have shown that [Ru(η6-p-cym)(saha)Cl] has moderate cytotoxicity (IC50 = 85 μM) against two ovarian cancer cell lines, namely A2780 and A2780 cisR.10
However, significantly less is known about the corresponding osmium complexes with the [OsII(η6-arene)] entity. Half-sandwich osmium(II) compounds with (N,N)11–14 (N,O)15,16 or (O,O)11,17 chelating ligands have been synthesized and tested against different cancer cell lines. In particular, acetylacetonato11 or maltolato17 containing complexes, [Os(η6-p-cym)(O,O)Cl], were found to be capable of fast ligand exchange in aqueous solution, resulting in the formation of an inactive hydroxo species at physiological pH. In the case of hydroxamates as (O,O) donors interacting with any forms of Os, only one report was found in the literature. Here, a reaction of [OsII(bpy)2Br2] (bpy = 2,2′-bipyridine) with N-arylbenzohydroxamic acids produced a cyclometalate of Os(III). The benzanilide which was formed from the hydroxamate coordinated as a dianionic C,N-donor.18 To our knowledge, there are no reports on Os complexes with (O,O) coordinated hydroxamate in the literature.
A comparison of the rate of ligand exchange reactions in half-sandwich Ru and Os complexes, [M(η6-arene)(XY)Z], indicates that the rate is 3–5 orders of magnitude smaller in Os complexes than in their corresponding ruthenium analogues.11 In general, while (N,N) donor ligand containing Ru complexes typically have suitable kinetic inertness and therefore high antiproliferative activity, the corresponding Os complexes might be less active due to the above kinetic differences. On the other hand, while (O,O) chelated Ru complexes can be too labile, the Os analogues might be of the appropriate kinetic behaviour and thus exert greater biological activity. Deprotonation of the water molecule after replacing Z auxiliary ligand in aqueous solution may also result in the formation of an inactive hydroxo species, and this is more pronounced for Os than Ru complexes with a given XY chelator.17 Since strongly coordinating XY are capable of shifting this process above pH 7.4, it is possible that Os-hydroxamates might yield good potential drug candidates.
Recently we have found that reaction of [Ru(η6-p-cym)Cl2]2 with benzohydroxamate (Chart 1) in the presence of non-coordinating triflate counter ion produced a dinuclear complex in which two half-sandwich metal centers were linked together with two hydroxamates. In this structure the carbonyl oxygens of the bha ligands coordinate to one of the Ru units, and the hydroxamate oxygens bridge to the two Ru atoms.8 In contrast, spectroscopic and MS results suggest that the presence of other ligands (e.g.Cl−) allows monodentate coordination yielding monomeric [Ru(η6-p-cym)(ha)Cl] (ha = hydroxamate) type species in solution.8
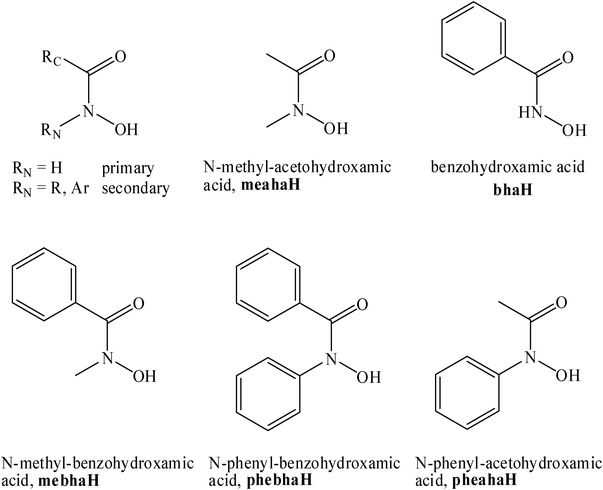 | ||
| Chart 1 | ||
In the current study, we sought to gain deeper insight into the effect of the size of the metal ion (Ruvs. Os), the steric demand of the RC and RN substituents at the hydroxamate group, the effect of the coordination of primary (RN = H) or secondary (RN = alkyl or aryl) hydroxamate (Chart 1) and the role of any monodentate ligand (Cl−, pyridine) may exert on the stability, stoichiometry, nuclearity and binding architecture of the half-sandwich metal(II) hydroxamate complexes. Herein we report the syntheses, solid state characterization and X-ray structures of a series osmium and ruthenium hydroxamate complexes, together with their in vitro anti-cancer potential using human ovarian cancer cell lines (A2780 and A2780cisR).
2. Experimental
2.1. Starting materials
OsO4, RuCl3·xH2O, α-terpinene, N-methylhydroxylamine hydrochloride, AgCF3SO3, acetyl chloride, benzoyl chloride, nitrobenzene, pyridine, sodium methoxide, benzohydroxamic acid, [Pt(NH3)2Cl2] were commercial products of the highest purity available (Heraeus, Aldrich, Merck or Fluka), and used as received. Solvents were dried and distilled according to standard methods.19N-phenylhydroxylamine hydrochloride was prepared from nitrobenzene while N-methyl-acetohydroxamic, N-phenyl-acetohydroxamic and N-phenyl-benzohydroxamic acid were prepared from acetyl or benzoyl chloride, by reaction with N-methyl- or N-phenylhydroxylamine following literature methods.20,21H2[OsCl6] was obtained as an orange-red viscous oil after refluxing OsO4 in an excess of concentrated aqueous HCl for 40 h, and removing any liquid by rotary evaporation under reduced pressure.22[Ru(η6-p-cym)Cl2]2 and [Ru(η6-p-cym)(acetone)3](CF3SO3)2 were synthesized and purified according to literature methods.23,24[Os(η6-p-cym)Cl2]2 was prepared by modification of a previously published procedure.25[Os(η6-p-cym)(acetone)3](CF3SO3)2 and [Os(η6-p-cym)(methanol)3](CF3SO3)2 were obtained in a similar manner as [Ru(η6-p-cym)(acetone)3](CF3SO3)2.24 H-NMR spectra were recorded on a Bruker AM360 FT-NMR instrument at room temperature in (CD3)2SO, and referenced to TMS as the internal standard. IR spectra (KBr pellets) were recorded on a Perkin Elmer FTIR Paragon 1000 PC instrument, and ESI-MS spectra (methanolic solutions) with a Bruker micrOTOF-Q 9 instrument in the positive mode. Elemental analyses (C, H, N, S) were conducted on an Elementar Variomicro Cube instrument at the Department of Organic Chemistry, Debrecen University, Hungary. DMF, all cell culture reagents and media were purchased from Sigma-Aldrich Ireland, Ltd, unless otherwise stated.2.2. Crystal structure analysis
Diffraction intensity data collection was carried out on a Bruker–Nonius MACH3 or an Agilent–Oxford SuperNova diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by the SIR-92 program26 and refined by full-matrix least-squares method on F2, with all non-hydrogen atoms refined with anisotropic thermal parameters using the SHELXL-97 package;27 publication material was prepared with the WINGX- suite.28 All hydrogen atoms were located geometrically, and refined using the riding model. Crystallographic and experimental details are summarized in Table 1.| [Ru(η6-p-cym)(meaha)]2(CF3SO3)2 | [Os(η6-p-cym)(meaha)Cl] | [Ru(η6-p-cym)(phebha)Cl] | [{Os(η6-p-cym)(meaha)}(μ-O){OsO(meaha)2}]Cl | [Ru(η6-p-cym)(bha)py](CF3SO3) | [Ru(η6-p-cym)(phebha)py](CF3SO3) | [Os(η6-p-cym)(μ–OH)py]2(CF3SO3)2 | |
|---|---|---|---|---|---|---|---|
| Compound | 5 | 6 | 9 | 17 | 12 | 14 | 16 |
| Chemical formula | C28H40F6N2O10Ru2S2 | C13H20ClNO2Os | C23H24ClNO2Ru | C20H32F3N3O11Os2S | C23H25F3N2O5RuS | C29H29F3N2O5RuS | C32H40F6N2O8Os2S2 |
| Formula weight/g mol−1 | 944.88 | 447.95 | 482.95 | 959.95 | 599.58 | 675.67 | 1139.18 |
| Crystal system | Triclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Tetragonal | Orthorhombic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P21/c | Pc21n | P21/c | P21/n | P 42bc | Pbca |
| a/Å | 9.337 (1) | 16.0879 (6) | 9.902 (3) | 14.2845 (3) | 17.902 (1) | 17.482(5) | 10.209 (5) |
| b/Å | 12.272 (1) | 10.0754 (4) | 10.380 (1) | 9.4565 (2) | 14.938 (1) | 17.482(5) | 17.512 (5) |
| c/Å | 17.763 (1) | 17.8788 (6) | 21.015 (1) | 21.8716 (4) | 19.641 (1) | 19.478(5) | 21.043 (5) |
| α (°) | 99.83 (1)° | 90 | 90 | 90 | 90 | 90 | 90 |
| β (°) | 103.22 (1)° | 100.93 (3)° | 90 | 105.51 (2)° | 105.36 (1)° | 90 | 90 |
| γ (°) | 105.48 (1)° | 90 | 90 | 90 | 90 | 90 | 90 |
| V Å−1³ | 1850.3 (3) | 2845.40 (18) | 2160.0 (7) | 2846.84 (10) | 5064.8 (5) | 5953(3) | 3762 (2) |
| Z | 2 | 8 | 4 | 4 | 8 | 8 | 4 |
| D calc/Mgm-3 | 1.696 | 2.091 | 1.485 | 2.247 | 1.573 | 1.508 | 2.008 |
| μ(Mo-Kα)/mm−1 | 1.01 | 9.14 | 0.87 | 9.07 | 0.76 | 0.66 | 6.94 |
| Crystal color/morphology | orange/prism | purple/block | yellow/prism | brown/prism | yellow/prism | orange/prism | yellow/prism |
| Crystal size | 0.25 × 0.2 × 0.12 | 0.18 × 0.16 × 0.06 | 0.3 × 0.25 × 0.15 | 0.3 × 0.13 × 0.1 | 0.3 × 0.25 × 0.24 | 0.3 × 0.25 × 0.2 | 0.45 × 0.3 × 0.25 |
| T/K | 293 | 150 | 293 | 150 | 293 | 293 | 293 |
| R int /% | 8.1 | 4.3 | 1.4 | 3.6 | 2.2 | 6.7 | 0.0 |
| Reflections: collected | 7046 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 078 078 |
2287 | 22172 |
9943 | 7558 | 3409 |
| Reflections unique | 6618 | 5185 | 2203 | 5202 | 9360 | 3011 | 3409 |
| Reflections observed, I > 2σ(I) | 4812 | 4003 | 1493 | 4387 | 6201 | 1909 | 2407 |
| No. of parameters | 534 | 289 | 256 | 370 | 643 | 373 | 241 |
| R[F2 > 2σ(F2)]/% | 0.058 | 0.028 | 0.065 | 0.018 | 0.098 | 0.052 | 0.050 |
| wR(F2)/% | 0.170 | 0.05 | 0.113 | 0.042 | 0.239 | 0.129 | 0.222 |
| S | 1.11 | 0.87 | 1.12 | 0.96 | 1.09 | 1.04 | 1.09 |
2.3. Synthesis of complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1604 s (C–C), 1412 s, 1274 vs. (triflate), 1260 vs. (triflate), 1158 s, 1030 s (triflate), 886 s, 638 s (triflate). MS (ESI-TOF): m/z (%) = 414.112 (100) [M–2CF3SO3]2+.
O), 1472 w, 1437 w, 1263 vs. (triflate), 1227 vs. (triflate), 1165 s, 1033 vs. (S–O), 640 w, 518 w (triflate). MS (ESI-TOF): m/z (%) = 324.058 (100) [M–2CF3SO3]2+.
O), 1468 s, 1434 s, 1162 m, 752 s, 654 s, 592 s. MS (ESI-TOF): m/z (%) = 414.113 (100) [M–Cl]+.
O), 1468 s, 1432 s, 954 m, 752 s 652 s, 568 s. MS (ESI-TOF): m/z (%) = 324.058 (100) [M–Cl]+.
O), 1450 vs. (py), 1284 vs, 1226 vs. (triflate), 1150 vs, 1030 vs. (S–O), 754 vs, 702 vs. (py), 636 vs, 518 s (triflate). MS (ESI-TOF): m/z (%) = 414.116 (100) [M–CF3SO3–Py]+.
O), 1470 s, 1448 s (py), 1424 s, 1404 m, 1276 vs. (triflate), 1224 s (triflate), 1030 vs. (triflate), 952 m, 752 s, 638 vs, 572 s, 518 m (triflate). MS (ESI-TOF): m/z (%) = 324.058 (100) [M–CF3SO3–Py]+.
O), 1507, 1481, 1448 (py), 1294, 1224 (triflate), 1159, 1028 (triflate), 913, 763, 694 (triflate), 637, 567, 515 (triflate). MS (ESI-TOF): m/z (%) = 372.053 (100) [M–CF3SO3–Py]+.
O), 1432 vs, 1274 vs. (triflate), 1146 vs, 1031 vs. (triflate), 772 vs. (py), 696 vs, 638 vs, 594 s, 516 s (triflate), 446 m. MS (ESI-TOF): m/z (%) = 538,147 (100) [M–CF3SO3–Py]+.
O), 1448 vs. (py), 1156 w, 1064 m (py), 770 vs. (py), 750 m, 700 m (py), 594 m. MS (ESI-TOF): m/z (%) = 414.117 (100) [M–Cl–Py]+, 412.114 (80) [M–Cl–Py]+.
O), 1604 s (C–C), 1412 s, 1274 vs. (triflate), 1260 vs. (triflate), 1158 s, 1030 s (triflate), 886 s, 638 s (triflate). MS (ESI-TOF): m/z (%) = 414.112 (100) [M–2CF3SO3]2+.
O), 1472 w, 1437 w, 1263 vs. (triflate), 1227 vs. (triflate), 1165 s, 1033 vs. (S–O), 640 w, 518 w (triflate). MS (ESI-TOF): m/z (%) = 324.058 (100) [M–2CF3SO3]2+.
O), 1468 s, 1434 s, 1162 m, 752 s, 654 s, 592 s. MS (ESI-TOF): m/z (%) = 414.113 (100) [M–Cl]+.
O), 1468 s, 1432 s, 954 m, 752 s 652 s, 568 s. MS (ESI-TOF): m/z (%) = 324.058 (100) [M–Cl]+.
O), 1450 vs. (py), 1284 vs, 1226 vs. (triflate), 1150 vs, 1030 vs. (S–O), 754 vs, 702 vs. (py), 636 vs, 518 s (triflate). MS (ESI-TOF): m/z (%) = 414.116 (100) [M–CF3SO3–Py]+.
O), 1470 s, 1448 s (py), 1424 s, 1404 m, 1276 vs. (triflate), 1224 s (triflate), 1030 vs. (triflate), 952 m, 752 s, 638 vs, 572 s, 518 m (triflate). MS (ESI-TOF): m/z (%) = 324.058 (100) [M–CF3SO3–Py]+.
O), 1507, 1481, 1448 (py), 1294, 1224 (triflate), 1159, 1028 (triflate), 913, 763, 694 (triflate), 637, 567, 515 (triflate). MS (ESI-TOF): m/z (%) = 372.053 (100) [M–CF3SO3–Py]+.
O), 1432 vs, 1274 vs. (triflate), 1146 vs, 1031 vs. (triflate), 772 vs. (py), 696 vs, 638 vs, 594 s, 516 s (triflate), 446 m. MS (ESI-TOF): m/z (%) = 538,147 (100) [M–CF3SO3–Py]+.
O), 1448 vs. (py), 1156 w, 1064 m (py), 770 vs. (py), 750 m, 700 m (py), 594 m. MS (ESI-TOF): m/z (%) = 414.117 (100) [M–Cl–Py]+, 412.114 (80) [M–Cl–Py]+.
2.4. Cell lines and cell culture
The in vitro anti-cancer chemotherapeutic potential of test compounds was determined using two human-derived malignant ovarian cancer cell lines (A2780 and A2780cisR). Both cell lines were a kind gift from Dr Maria Morgan, Dept. of Molecular & Cellular Therapeutics, Royal College of Surgeons, Ireland, Dublin. A2780cisR cells are a cisplatin resistant human ovarian cell line developed by chronic exposure of the parent cisplatin sensitive A2780 cell line to increasing concentrations of cisplatin. Furthermore, these cells are cross-resistant to melphalan, adriamycin and irradiation. This resistant phenotype was maintained by pulsing cells during every third passage with cisplatin (1 μM). Both cell lines were maintained in RPMI-1640 media with Earle's balanced salt solution (EBSS) containing 1.5 g L−1sodium bicarbonate, 2 mM L-glutamine, 100 U ml−1penicillin, 100 μg ml−1streptomycin and 10% (v/v) foetal bovine serum (FBS). These two model cell lines were grown at 37 °C in a humidified atmosphere with 5% CO2 and were in the exponential phase of growth at the time of inclusion in cytotoxicity assays.2.5. Assessment of cytotoxicity, using MTT assay
Each of the two cell lines (100 μl) were seeded at a density of 2.5 × 104cells cm−3 into sterile 96 well flat-bottomed plates (Sarstedt) and grown in 5% CO2 at 37 °C. Test compounds were dissolved in DMF and diluted with culture media. The maximum percentage of DMF present in all wells was 0.5% (v/v). Solutions (100 μl) of complexes 1, 5–8, 10 and 14 were added to replicate wells in the concentration range of 10–200 μM and incubated for 72 h. A miniaturised viability assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was carried out according to the method described by Mosmann.29 The IC50 value, defined as the drug concentration causing a 50% reduction in cellular proliferation, was calculated for each complex. Each assay was carried out using five replicates and repeated on at least three separate occasions. Proliferation was calculated as a percentage of solvent-treated control cells, and expressed as a percentage of control. The significance of any reduction in cellular viability was determined using one-way ANOVA (analysis of variance). A probability of 0.05 or less was deemed statistically significant.3. Results and discussion
3.1. Synthesis and spectroscopic characterization
[M(η6-p-cym)(ha)]2(CF3SO3)2 (4), [M(η6-p-cym)(ha)Cl] (6) or [M(η6-p-cym)(ha)(py)]X (10) (M = Os, Ru; ha = monohydroxamate, py = pyridine, X = Cl− or CF3SO3−) type complexes were prepared in moderate to good yields from [M(η6-p-cym)Cl2]2 precursors or from the coordinative unsaturated 2–3, respectively, by reacting them with the corresponding hydroxamates (see Chart 1) and with pyridine for 10–15 at room temperature. The novel compounds were air stable crystalline solids, and were soluble in polar solvents such as dichloromethane, acetone, dimethylsulfoxide, methanol and also water.1H-NMR spectra of the complexes showed the expected resonance signals; and a representative sample spectrum of 5 is presented in Fig. S1, ESI.† Complexation resulted in downfield shifts (0.05–0.11 ppm) of the p-cymene ring protons in 4–5 but highfield shifts (0.18–0.33 ppm) for 6–9 when the corresponding resonances with those in the [M(η6-p-cym)Cl2]2 precursors were compared. This difference can be explained by an electron donating effect of the negatively charged chloride ion in the coordination sphere beside the hydroxamate in 6–9. Similarly, as a result of coordination of the hydroxamate (0.02–0.61 ppm) or pyridine (0.10–0.40 ppm) to the half-sandwich metal center, downfield shifts of the ligand protons are detected relative to the uncomplexed ligands except for the N2 and N6 protons of the pyridine ring which show highfield shifts. Furthermore, [M(η6-p-cym)(ha)(py)]X type complexes with the same ligands in the coordination sphere of the metal ion (e.g.10vs.15), show practically identical chemical shift values, regardless of the X counter ion.
The NMR spectrum of 16 indicated that three different species were formed in d6-DMSO solution after sample preparation using the solid crystalline complex. Selected parts of the spectrum are shown in Fig. S2, ESI.†DOSY experiments proved that besides free pyridine (D, Fig. S2), two other pyridine-containing complexes were present in solution, with three sets of signals belonging to the p-cymene protons being identifiable. Furthermore, three new singlets were also present in the spectrum which most probably belong to the hydroxide groups of the complexes. Complexes with the M(μ-OH)M or M(μ-OH)2M motif showed signals attributed to bridging OH in the 3.0–4.7 ppm range.30–32 Therefore, the data above may suggest that after dissolution of 16, partial or complete dissociation of the coordinated pyridine ligands occurred and besides free pyridine, it is possible that intact [Os(η6-p-cym)(μ-OH)(py)]22+, [Os(η6-p-cym)(py)(μ-OH)2Os(η6-p-cym)]2+ and [Os(η6-p-cym)(μ-OH)]22+ ions are also present.
IR spectra of the novel hydroxamate complexes exhibited a new sharp band in the range 1545–1645 cm−1 compared to those of [M(η6-p-cym)Cl2]2 precursors which was assigned to the νCO of the coordinating hydroxamates. Comparison of the corresponding Ru and Os complexes indicated that these stretches appeared at slightly smaller wavenumbers for the former metal ion. The presence of the half-sandwich M(II) core was revealed by the characteristic five sharp bands in the wavenumber range of 3100–2850 cm−1.8 The triflate counter ion was indicated by the characteristic stretches at 1274, 1260, 1226, 1030 and 518 cm−1,33 while coordinated pyridine was identified at 1448, 1065, 806, 771 and 701 cm−1.
Electrospray ionization mass spectrometric (ESI-MS) analysis in the positive mode provided further proof for the identity of the complexes. As found previously,8 the ESI-MS conditions produced no difference in the spectra to that of the corresponding triflate or chloride containing complexes (e.g.4vs.6 or 5vs.7), regarding the major peaks. The same was observed with the [M(η6-p-cym)(ha)(py)]X complexes, revealing that chloride ion and pyridine dissociate at the ESI-MS conditions applied. All the mass spectra displayed the correct isotopic pattern.
3.2. X-ray crystallographic studies
Previously we have shown that in the presence of weakly coordinating counter ion like triflate, the primary (RN = H) hydroxamate ligand, bha− (Chart 1) was capable of bridging two half-sandwich [Ru(η6-p-cym)]2+ units with the occupation of all coordination sites.8 Our recent results demonstrate that a secondary (RN = CH3) hydroxamate, meaha− (Chart 1), binds to the metal ion in an identical manner. The ORTEP diagram of [Ru(η6-p-cym)(μ-meaha)]22+ (5) shown in Fig. 1. with key bond distances and angles appearing in the caption of Fig. 1. Regarding the Ru–O distances, the bridging Ru–O bonds [2.113(5)–2.159(5) Å] were significantly longer than those of the Ru-carbonyl O bonds [2.065(5) Å, 2.075(5) Å]. Comparison of the corresponding data with those determined previously8 for [Ru(η6-p-cym)(μ-bha)]22+ indicated no significant differences in the geometry and distances in the two dinuclear structures to that of the primary or secondary hydroxamate. However, with the appropriate Os precursors, we were able to obtain a pure crystalline solid only with secondary hydroxamates (RN = alkyl, aryl). This difference between the [M(η6-p-cym)]2+ (M = Ru, Os) cores can be explained by the 5dosmium(II) being more easily involved in redox reactions.34 In particular, redox reactions with the oxidation of the metal ion by primary hydroxamates (RN = H) yielding amide has been well documented in the literature for Fe(II) or VO(IV).35,36![X-Ray structure of the cation of [Ru(η6-p-cym)(μ-meaha)]2(CF3SO3)2 (5). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Ru(1) –O(1) 2.113(5), Ru(1) –O(2) 2.065(5), Ru(1) –O(11) 2.151(5), Ru(2) –O(1) 2.160(5), Ru(2) –O(12) 2.071(5), Ru(2) –O(11) 2.112(5), Ru–Carene(avr.) 2.170(10); O(1) –Ru(1) –O(2) 76.8(2), O(1) –Ru(1) –O(11) 77.9(2), O(2) –Ru(1) –O(11) 88.3(2), O(11) –Ru(2) –O(12) 76.3(2), O(11) –Ru(2) –O(1) 77.7(2), O(12) –Ru(2) –O(1) 87.3(2), Ru(1) –O(1) –Ru(2) 97.6(2), Ru(1) –O(11) –Ru(2) 97.9(2).](/image/article/2012/RA/c1ra00998b/c1ra00998b-f1.gif) | ||
| Fig. 1 X-Ray structure of the cation of [Ru(η6-p-cym)(μ-meaha)]2(CF3SO3)2 (5). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Ru(1) –O(1) 2.113(5), Ru(1) –O(2) 2.065(5), Ru(1) –O(11) 2.151(5), Ru(2) –O(1) 2.160(5), Ru(2) –O(12) 2.071(5), Ru(2) –O(11) 2.112(5), Ru–Carene(avr.) 2.170(10); O(1) –Ru(1) –O(2) 76.8(2), O(1) –Ru(1) –O(11) 77.9(2), O(2) –Ru(1) –O(11) 88.3(2), O(11) –Ru(2) –O(12) 76.3(2), O(11) –Ru(2) –O(1) 77.7(2), O(12) –Ru(2) –O(1) 87.3(2), Ru(1) –O(1) –Ru(2) 97.6(2), Ru(1) –O(11) –Ru(2) 97.9(2). | ||
The dinuclear complex formation was disfavoured in the presence of chloride ions which are capable of stronger interaction with the half-sandwich metal cores. As demonstrated with both metals, and with different hydroxamates, two of the coordination sites of the [M(η6-p-cym)]2+ cores are taken by a hydroxamate (O,O) chelate, while chloride was present at the third position. A representative example of the ORTEP structure of [Os(η6-p-cym)(meaha)Cl] is shown is Fig. 2, while the structure of [Ru(η6-p-cym)(phebha)Cl] in Fig. S3, ESI.† The corresponding bond distances and angles (captions to Fig. 2 and S3) revealed no significant differences when the two metals or the different type hydroxamate ligands were compared. Both the Os–Cl (2.425 Å) and the Ru–Cl (2.411 Å) distances were shown to be in the expected range (Os: 2.40(7), Ru: 2.42(6) Å).37
![X-Ray structure of [Os(η6-p-cym)(meaha)Cl] (6). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Os(1) –Cl(1) 2.429(2), Os(1) –O(1) 2.075(4), Os(1) –O(2) 2.092(4), Os(1) –Carene(avr.) 2.172(25); O(1) –Os(1) –O(2) 76.68(15), Cl(1) –Os(1) –O(1) 82.19(11), Cl(1) –Os(1) –O(2) 82.99(11).](/image/article/2012/RA/c1ra00998b/c1ra00998b-f2.gif) | ||
| Fig. 2 X-Ray structure of [Os(η6-p-cym)(meaha)Cl] (6). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Os(1) –Cl(1) 2.429(2), Os(1) –O(1) 2.075(4), Os(1) –O(2) 2.092(4), Os(1) –Carene(avr.) 2.172(25); O(1) –Os(1) –O(2) 76.68(15), Cl(1) –Os(1) –O(1) 82.19(11), Cl(1) –Os(1) –O(2) 82.99(11). | ||
Unexpectedly, we have found the formation of an unusual dinuclear Os complex (17) with the partial oxidation of the metal ion, when a solution of [Os(η6-p-cym)(meaha)]2(CF3SO3)2 (4) in a mixture of acetone/hexane, was left to evaporate slowly under aerobic conditions, and at room temperature in a narrow crystallization tube. The X-ray structure of the isolated yellow solid is presented in Fig. 3, while the key bond angles and distances are summarized in the caption to Fig. 3. It can be seen that the new dinuclear complex consisted of a half-sandwich [Os(η6-p-cym)] core to which a meaha− ligand is bound via (O,O) chelate. This unit was linked via an oxo group to another metal core in which an osmium with octahedral geometry was located. The remaining coordination sites of the second Os were occupied by two (O,O) chelates of two meaha− ligands and an oxo group. Comparison of the Os(2)–O(3) (2.035 Å), Os(1)–O(3) (1.781 Å) distances and the Os(1)–O(3)–Os(2) bond angle (162°) with those of dinuclear oxo bridged Os complexes38,39 also supports the theory that an oxo group, and not a hydroxide ion, binds the two metal cores together. While octahedral Os complexes with an Os–O single bond feature 2.09(6) Å while with an OsO double bond 1.73(3) Å distances37 the short value of Os(1)–(O4) (1.718 Å) in 17 is indicative for an OsO bond. As the complex also contains a non-coordinating chloride ion the second osmium should have a +6 oxidation state in 17. Similar oxocationic octahedral osmium complexes with the OsO4+ core have already been reported in the literature with (O,O) ligands.40–42
![X-Ray structure of [{Os(η6-p-cym)(meaha)}(μ–O){OsO(meaha)2}]CF3SO3 (17). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Os(1) –O(1) 1.979(2), Os(1) –O(2) 2.054(2), Os(1) –O(3) 1.781(2), Os(1) –O(4) 1.718(3), Os(1) –O(11) 1.977(3), Os(1) –O(12) 2.058(3), Os(2) –O(3) 2.035(2)Os(2) –O(21) 2.063(3), Os(2) –O(22) 2.058(2), Os(1) –Carene(avr.) 2.178(14); Os(1) –O(3) –Os(2) 162.58(14), O(21) –Os(2) –O(22) 78.07(10), O(21) –Os(2) –O(3) 82.51(10), O(22) –Os(2) –O(3) 81.74(10), O(3) –Os(1) –O(4) 169.84(13).](/image/article/2012/RA/c1ra00998b/c1ra00998b-f3.gif) | ||
| Fig. 3 X-Ray structure of [{Os(η6-p-cym)(meaha)}(μ–O){OsO(meaha)2}]CF3SO3 (17). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Os(1) –O(1) 1.979(2), Os(1) –O(2) 2.054(2), Os(1) –O(3) 1.781(2), Os(1) –O(4) 1.718(3), Os(1) –O(11) 1.977(3), Os(1) –O(12) 2.058(3), Os(2) –O(3) 2.035(2)Os(2) –O(21) 2.063(3), Os(2) –O(22) 2.058(2), Os(1) –Carene(avr.) 2.178(14); Os(1) –O(3) –Os(2) 162.58(14), O(21) –Os(2) –O(22) 78.07(10), O(21) –Os(2) –O(3) 82.51(10), O(22) –Os(2) –O(3) 81.74(10), O(3) –Os(1) –O(4) 169.84(13). | ||
Dinuclear complex formation can also be hindered if other monodentate ligands capable of relatively strong coordination to the half-sandwich metal cores (e.g.pyridine) are present beside the coordinating hydroxamate. As an example, the crystal structure of [Ru(η6-p-cym)(bha)(py)]CF3SO3 (12) appears in Fig. 4, while that of [Ru(η6-p-cym)(phebha)(py)]CF3SO3 (14) is shown in Fig. S4, ESI.† In both structures, beside the hydroxamate (O,O) chelate, a pyridine N can be found in the third coordination site of the metal ion. 14 is also stabilized by hydrogen bonds which can be detected between the hydroxamate NH's as donors and triflate O's as acceptors. The dimensions of these H bonds are as follows: N(1)…O(81) = 2.799(13) Å, H(N1)…O(81) = 1.98(4) Å, N(1)–H(N1)…O(81) = 160(10)° and N(11)…O(73i) = 2.891(12) Å, H(N11)…O(73i) = 2.13(6) Å, N(11)–H(N11)…O(73i) = 148(9)°. Comparison of the corresponding bond length and angle values (captions to Fig. 4 and S4) indicates that benzohydroxamate (RC = Phe, RN = H) and N-phenyl-benzohydroxamate (RC = RN = Phe), the latter with larger steric demand, may behave very similarly in these mixed complexes.
![X-Ray structure of [Ru(η6-p-cym)(bha)(py)]CF3SO3 (12). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Ru(1) –O(1) 2.044(7), Ru(1) –O(2) 2.084(6), Ru(1) –N(10) 2.123(8), Ru(1) –Carene(avr.) 2.169(23); O(1) –Ru(1) –O(2) 78.3(3), N(10) –Ru(1) –O(1) 83.1(3), N(10) –Ru(1) –O(2) 82.1(3).](/image/article/2012/RA/c1ra00998b/c1ra00998b-f4.gif) | ||
| Fig. 4 X-Ray structure of [Ru(η6-p-cym)(bha)(py)]CF3SO3 (12). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Ru(1) –O(1) 2.044(7), Ru(1) –O(2) 2.084(6), Ru(1) –N(10) 2.123(8), Ru(1) –Carene(avr.) 2.169(23); O(1) –Ru(1) –O(2) 78.3(3), N(10) –Ru(1) –O(1) 83.1(3), N(10) –Ru(1) –O(2) 82.1(3). | ||
Although we do not have an X-ray structure of 15 with the meaha− ligand, the obtained analytical data are consistent with its stoichiometry. At the same time, during the synthesis of the analogous Os complex with mebhaH, we were unsuccessful in obtaining [Os(η6-p-cym)(mebha)(py)]CF3SO3. Instead, using 20 h reaction time and after work-up of the reaction mixture, a novel dihydroxo bridged mixed pyridine complex, 16, could be isolated and the X-ray structure determined. As Fig. 5 reveals, this symmetrical dinuclear Os complex has two half-sandwich [Os(η6-p-cym)(py)]+ units which are linked viahydroxide bridges. Key bond lengths and angles are summarized in the caption to Fig. 5. Os–OH distances (2.088(9) and 2.091(9) Å) in 16 are in the range similar to that of Os complexes, showing an average Os–OH distance37 of 2.11(5) Å for the published four structures having hydroxo bridges between the two osmium atoms. The appropriate data for 16 is also in good agreement with the published Os–O(H)–Os angles (average: 103°) and Os–Os distances, average of 3.23(7) Å. Formation of 16 is likely due to the decomposition of the mebha− ligand, and to the formation of kinetically inert hydroxo bridged species, with the involvement of trace water present during the reaction or subsequent work-up.
![X-Ray structure of [Os(η6-p-cym)(μ–OH)(py)]2(CF3SO3)2 (16). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Os(1) –O(1) 2.091(9), Os(1) –O(1_i) 2.088(9), Os(1_i) –O(1) 2.088(9), Os(1) –N(21) 2.104(11), Os(1) –Carene(avr.) 2.182(20); Os(1) –O(1) –Os(1_i) 107.3(4), O(1) –Os(1) –N(21) 84.6(4), O(1) –Os(1) –O(1_i) 72.7(4), O(1_i) –Os(1) –N(21) 81.5(4).](/image/article/2012/RA/c1ra00998b/c1ra00998b-f5.gif) | ||
| Fig. 5 X-Ray structure of [Os(η6-p-cym)(μ–OH)(py)]2(CF3SO3)2 (16). Thermal ellipsoids show 50% probability with partial numbering scheme. Selected bond lengths (Å) and angles (°): Os(1) –O(1) 2.091(9), Os(1) –O(1_i) 2.088(9), Os(1_i) –O(1) 2.088(9), Os(1) –N(21) 2.104(11), Os(1) –Carene(avr.) 2.182(20); Os(1) –O(1) –Os(1_i) 107.3(4), O(1) –Os(1) –N(21) 84.6(4), O(1) –Os(1) –O(1_i) 72.7(4), O(1_i) –Os(1) –N(21) 81.5(4). | ||
3.3. Cytotoxicity in cancer cell lines
The in vitro anti-cancer chemotherapeutic potential of this series of Ru- and Os-hydroxamic acid complexes was determined using two human-derived ovarian cancer cell lines; a parental cell line (A2780) and a cisplatin-resistant variant (A2780 cisR). The results show that all the complexes (1) can be regarded as inactive (IC50 > 200 μM), since they failed to reduce the viability of either model cell line across both the concentration range (0–200 μM) and incubation period (72 h) studied. In contrast, cisplatin displayed a significant reduction in cellular viability toward both cell lines, with mean IC50 values of 1.3 ± 0.1 and 9.7 ± 1.0 μM seen in A2780 and A2780 cisR cells, respectively.4. Conclusions
The results of this paper showed that secondary monohydroxamates (RN = alkyl, aryl) were capable of the double bridging of two half-sandwich M(II) (M = Ru, Os) cores, and the same holds true for the primary hydroxamate (RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) H) complexes of Ru. The X-ray crystal structure of 5 demonstrated that in the dinuclear entity, the two meaha− ligands were bound in an identical manner via deprotonated O's as bridging atoms and carbonyl O's as monodentate atoms. As [Os(η6-p-cym)]2+ was less resistant to redox reactions, it was not possible to isolate stable complexes with primary hydroxamates. The presence of monodentate co-ligands resulted in the formation of mononuclear complexes with the expected [M(η6-p-cym)(ha)X]n+ (M = Os, Ru; ha = hydroxamate, X = py, Cl−) stoichiometry, with [Os(η6-p-cym)(meaha)Cl] (6), being the first organometallic Os(II)-hydroxamate characterized by X-ray crystallography. The unexpected formation of an oxo bridged dinuclear OsVI/OsII complex, 17, consisting an octahedral Os(VI) core and an intact half-sandwich [Os(η6-p-cym)]2+ unit suggests that partial oxidation of the [Os(η6-p-cym)]2+ core may happen under aerobic conditions.
H) complexes of Ru. The X-ray crystal structure of 5 demonstrated that in the dinuclear entity, the two meaha− ligands were bound in an identical manner via deprotonated O's as bridging atoms and carbonyl O's as monodentate atoms. As [Os(η6-p-cym)]2+ was less resistant to redox reactions, it was not possible to isolate stable complexes with primary hydroxamates. The presence of monodentate co-ligands resulted in the formation of mononuclear complexes with the expected [M(η6-p-cym)(ha)X]n+ (M = Os, Ru; ha = hydroxamate, X = py, Cl−) stoichiometry, with [Os(η6-p-cym)(meaha)Cl] (6), being the first organometallic Os(II)-hydroxamate characterized by X-ray crystallography. The unexpected formation of an oxo bridged dinuclear OsVI/OsII complex, 17, consisting an octahedral Os(VI) core and an intact half-sandwich [Os(η6-p-cym)]2+ unit suggests that partial oxidation of the [Os(η6-p-cym)]2+ core may happen under aerobic conditions.
Complexes 1, 5–8, 10 and 14 were screened for possible in vitro anti-proliferative activity. Results showed that IC50 values were greater than 200 μM. This may be explained by fast ligand exchange reactions of the (O,O) donor hydroxamates. In the case of the Os complexes, it is possible that dissociation of administered compounds, may lead to the formation of less active species which are incapable of biological activity at low concentrations. However, given that similar complexes have previously shown antimicrobial activity, additional biological studies will focus on an assessment of their anti-bacterial (Gram positive and Gram negative) and anti-fungal profile.
Acknowledgements
We thank members of the EU COST Action D39 for motivating discussions. This work was supported by the Hungarian Scientific Research Fund (OTKA K76142), TAMOP 4.2.1./B-09/1/KONV-2010-0007 project co-financed by the European Union and the European Social Fund by the Technological Sector Research Programme, Strand III, under the European Social Fund and the Programme for Research in Third Level Institutes (2006–2010).References
- C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 3003 CrossRef CAS.
- P. A. Marks, Oncogene, 2007, 26, 1351 CrossRef CAS.
- P. A. Marks and W. S. Xu, J. Cell. Biochem., 2009, 107, 600 Search PubMed.
- M. Melchart, P. J. Sadler, in: Bioorganometallics, (ed. G. Jaouen), Wiley-VCH, 2006, p. 39–64.
- P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1 CAS.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and B. K. Keppler, in: Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, 2011, p. 160–164.
- I. Bratsos, D. Urankar, E. Zangrando, P. Genova-Kalou, J. Kosmrlj, E. Alessio and I. Turel, Dalton Trans., 2011, 40, 5188 RSC.
- P. Buglyó and E. Farkas, Dalton Trans., 2009, 8063 RSC.
- L. Bíró, E. Farkas and P. Buglyó, Dalton Trans., 2010, 39, 10272 RSC.
- P. Buglyó and D. Griffith, unpublished results.
- A. F. A. Peacock, A. Habtemariam, R. Fernández, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739 CrossRef CAS.
- A. F. A. Peacock, A. Habtemariam, S. A. Moggach, A. Prescimone, S. Parsons and P. J. Sadler, Inorg. Chem., 2007, 46, 4049 CrossRef CAS.
- L. K. Filak, G. Mühlgassner, M. A. Jakupec, P. Heffeter, W. Berger, V. B. Arion and B. K. Keppler, JBIC, J. Biol. Inorg. Chem., 2010, 15, 903 CrossRef CAS.
- L. K. Filak, G. Mühlgassner, F. Bacher, A. Roller, M. Galanski, M. A. Jakupec, B. K. Keppler and V. B. Arion, Organometallics, 2011, 30, 273 CrossRef CAS.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348 CrossRef CAS.
- R. Schuecker, R. O. John, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2008, 27, 6587 CrossRef CAS.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2007, 13, 2601 CrossRef CAS.
- A. Das, F. Basuli, L. R. Falvello and S. Bhattacharya, Inorg. Chem., 2001, 40, 4085 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 3rd Edition, Pergamon, Oxford, 1988.
- D. A. Brown, R. A. Geraty, J. D. Glennon and N. N. Choileain, Synth. Commun., 1985, 15, 1159 CAS.
- B. Monzyk and A. L. Crumbliss, J. Org. Chem., 1980, 45, 4670 CrossRef CAS.
- M. A. Bennett, T. W. Matheson, G. B. Robertson, A. K. Smith and P. A. Tucker, Inorg. Chem., 1980, 19, 1014 CrossRef CAS.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233 RSC.
- A. Schlüter, K. Bieber and W. S. Sheldrick, Inorg. Chim. Acta, 2002, 340, 35 CrossRef CAS.
- W. A. Kiel, R. G. Ball and W. A. G. Graham, J. Organomet. Chem., 1990, 383, 481 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- T. Mosmann, J. Immunol., 1983, 65, 55.
- D. Carmona, A. Mendoza, J. Ferrer, F. J. Lahoz and L. A. Oro, J. Organomet. Chem., 1992, 431, 87 CrossRef CAS.
- Q. F. Zang, R. D. Adams and W. H. Leung, Inorg. Chim. Acta, 2006, 359, 978 CrossRef CAS.
- M. Auzias, B. Therrien and G. Süss-Fink, Inorg. Chem. Commun., 2007, 10, 1239 Search PubMed.
- J. M. Alía, Y. Díaz de Mera, H. G. M. Edwards, F. J. García and E. E. Lawson, J. Mol. Struct., 1997, 408(409), 439–450 CrossRef.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, 5th Edition, Wiley, New York, 1988 Search PubMed.
- E. Farkas, É. A. Enyedy and I. Pócsi, J. Inorg. Biochem., 2004, 98, 1957 Search PubMed.
- P. Buglyó and N. Pótári, Polyhedron, 2005, 24, 837 CrossRef CAS.
- Cambridge Structural Database, Version 5.32 Update May, 2011.
- J. A. Christie, T. J. Collins, T. E. Krafft, B. D. Santarsiero and G. H. Spies, Chem. Commun., 1984, 198 RSC.
- C. L. Gross, J. L. Brumaghim and G. S. Girolami, Organometallics, 2007, 26, 2258 CrossRef CAS.
- T. Behling, M. V. Capparelli, A. C. Skapski and G. Wilkinson, Polyhedron, 1982, 1, 840 CrossRef.
- A. Struess and W. Preetz, Z. Naturforsch. B, 1998, 53, 823.
- S. Stanislas, A. L. Beauchamp and C. Reber, Inorg. Chem., 2000, 39, 2152 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 838351, 838352, 838353, 838354, 838355, 838356 and 838357. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00998b |
| This journal is © The Royal Society of Chemistry 2012 |