Cyclodextrin fibers via polymer-free electrospinning
Joshua L.
Manasco
,
Carl D.
Saquing†
,
Christina
Tang
and
Saad A.
Khan
*
Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695-7905. E-mail: khan@eos.ncsu.edu
First published on 14th February 2012
Abstract
Cyclodextrins (CDs) are intriguing amphiphilic molecules that consist of a hydrophilic outer structure and a hydrophobic core with the ability to act as hosts for both nonpolar and polar guests. Electrospinning is a facile yet effective method for producing non-woven mats of fibers with high aspect ratios. Cyclodextrin fibers would leverage the distinctive properties of these molecules with the unique properties of nanofibers. We report the fabrication of submicron hydroxypropyl-β-cyclodextrin (HPβCD) fibers from highly concentrated aqueous solutions by electrospinning without the addition of a carrier polymer. We focus on exploring solution properties that make fiber formation possible contrary to the widely accepted premise that molecular entanglement of macromolecules is required for electrospinning. The ability to electrospin these solutions is attributed to hydrogen-bonded aggregation between HPβCD molecules at high concentrations, as evidenced from an exponential increase in zero-shear viscosity and bound water as a function of concentration, as well as disruption of fiber formation upon addition of urea to the system.
1 Introduction
Cyclodextrin molecules (CDs) are doughnut-shaped cyclic oligosaccharides typically composed of 6, 7 or 8 member α-D-glucopyranose rings (α, β or γ CD, respectively). Native and functionalized CDs are used in various applications because of their ability to encapsulate moieties into their cavities.1–9 For example, CDs have been used as complexing agents or additives to aid in drug and flavor formulation.10–23 Alternatively, CDs have been used for the elimination of unwanted tastes and contaminants in consumer products.1 Further, CDs are used as catalysts for improved selectivity, and for separation and purification of industrial-scale products.2 In such applications, high specific surface area may lead to enhanced performance. Nanofibers have an intrinsically high specific surface area, and are thus desirable; and the smaller the fiber the larger the specific area.Electrospinning is a facile yet effective method for producing nanoscale non-woven mats of fibers by applying a high electric field to a polymer solution or jet.24–26 To date, electrospinning is generally limited to polymer solutions. CD containing nanofibers have been electrospun in multicomponent systems for various applications.27–37 The first reported use of CDs employed β-CD as a crosslinking agent to render nanofibers made of poly(acrylic acid) water insoluble.27 There are three cases where β-CD was employed in different capacities with poly(vinylpyrrollidone): as a carrier for bicomponent fibers, as a reducing/stabilizing agent for gold nanoparticles, and as a porogen to create pores in fibers.28–30 Recently Uyar et al. used α-CD to create pseudopolyrotaxanes inclusion complexes with poly(ethylene glycol) which they electrospun using poly(ethylene oxide) as a carrier polymer.31
We report the fabrication of fibers via electrospinning concentrated aqueous solutions of chemically modified cyclodextrin, HPβCD, the chemical structure of which is shown in Fig. 1. Notably, fibers were obtained in the absence of an electrospinnable polymer. This is contrary to the generally accepted paradigm that polymer entanglement is required for fiber formation 38–41 as CDs are small molecules. In the accepted paradigm, molecular entanglement that is achieved when the polymer concentration is well over the critical overlap concentration, c*, provides elasticity to the polymer solution and hence electrospinnability. However, Yu et al. showed that molecular entanglements is not a necessary condition for electrospinnability, but rather elasticity is the crucial property that determines fiber formation via electrospinning.42 They demonstrated this by electrospinning polymer solutions with concentrations below that where any entanglement transition is observed but had relaxation times longer than the time of extensional deformation. Subsequently, Talwar et al. also showed the importance of elasticity and relaxation time in electrospinnability.43 We report the electrospinnability of the CD molecule-water network that lacks entanglement as well as elasticity.
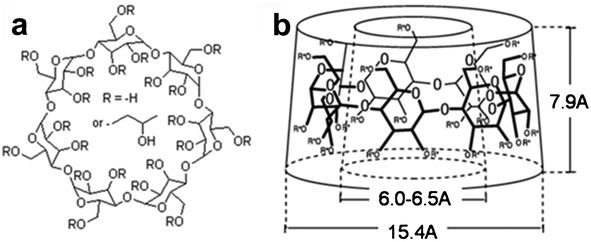 | ||
| Fig. 1 a) Chemical structure of hydroxypropyl β-cyclodextrin. b) Approximate dimensions of the toroid formed by β-cyclodextrin | ||
Recently, electrospinning has been extended beyond traditional polymers. For example, globular proteins have been electrospun. In this case, electrospinnability was attributed to disruption of the original tertiary structure (via trifluroethanol) and intramolecular disulfide bonds (via β-mercaptoethanol) which allowed for the formation of new inter- and intramolecular bonds.44 Low molecular weight compounds such as surfactants, specifically Gemini surfactants in water and methanol, have also been electrospun which was attributed to the presence of overlapping micelles.45 Additionally, fibers from phospholipids, namely lecithin in a mixture of chloroform and dimethylformamide, have also been reported due to the presence of entanglements between wormlike micelles.46 In parallel with our work, electrospinning of methyl-β-cyclodextrin in either dimethylformamide or water, as well as aqueous solutions of cyclodextrin inclusion complexes (hydroxypropyl-β-cyclodextrin and triclosan) have been reported possibly due to the entanglement of aggregates of the CD molecules.47,48 However, investigation of the role of solution properties affecting electrospinnability has yet to be fully explored.
We examine the role of solution properties known to affect electrospinnability, namely, conductivity, surface tension, and viscosity. Further, we use rheology and thermal analysis of the concentrated CD solutions to explore the formation of CD aggregates. Based on thermal analysis of bound water and rheological analysis we speculate that the CD aggregates that facilitate electrospinning form via a mechanism analogous to depletion flocculation. By investigating the effect of the addition of a chaotropic agent, we probe the nature of the intermolecular interactions leading to electrospinnability of this unique molecule.
2 Experimental
Materials
HPβCD (technical grade) was purchased from CTD, Inc (High Springs, FL) and used as received. The average degree of substitution for this derivative was 5.6 (computed mol. wt −1460 g mol−1). MβCD (average degree of substitution of ∼11.5 or ∼55%) 49 was generously donated by Wacker Chemie (Adrian, Michigan). The average degree of substitution for this derivative was 11.5 (computed mol. wt −1310 g mol−1). Urea was obtained from Sigma Aldrich and used as received. The HPβCD and MβCD solutions were made fresh by adding the respective amounts of CD to de-ionized water, and allowed to mix in a shaker bath overnight at 35 °C. All solutions were stored at 4 °C when not in use to prevent degradation.Solution characterization
Rheological experiments were performed at 25 °C using a TA Instruments AR-2000 stress controlled rheometer with a 4 cm, 2° cone and plate geometry. Measurements were performed at least thrice to ensure reproducibility within ± 5%. Surface tension measurements at the air/solution interface were made by the Wilhelmy plate method using a platinum rectangular thin blade. The electrical conductivity was measured using an EC meter (Fisher Accumet BASIC AB30).Bound water (not freezable) studies were done on the HPβCD solutions using a TA instruments® Q2000 Differential Scanning Calorimeter. Each sample (8–12 mg) was loaded into an aluminum hermetic pan and crimped. The samples were ramped down from room temperature to −60 °C and up to room temperature using a cooling/heating rate of 10 °C min−1. The amount of free water was calculated by measuring the melting energy of ice formed when the frozen solutions were melted and dividing the measured melting energy by the theoretical energy required for melting ice (333.55 J g−1). The energy of melting for the different HPβCD solutions was determined using the area of the melting peak.
Electrospinning
The electrospinning setup consisted of a grounded aluminum collector plate, a precision syringe pump (Harvard Apparatus, Holliston, MA) fitted with metallic needle (22 G) syringe, and a high-voltage power supply (Gamma High Voltage Research, model D-ES30 PN/M692 with a positive polarity). The tip-to-collector distance and solution flow rate were fixed at 12 cm and 0.5 mL h−1, respectively, and the applied voltage ranged between 5 and 11 kV.Fiber analysis
SEM measurements on gold coated fibers were obtained on a Hitachi S-3200 and FEI XL30 scanning electron microscope. The average fiber diameter was determined by measuring 100 individual fibers from multiple SEM images using Revolution SEM™ software (4Pi Analysis, Inc.) and Image J software (NIH). Nanofibers were deposited directly onto carbon-coated copper grids (Ted Pella) and analyzed using a FEI Tecnai G2 Twin transmission electrospin microscope (TEM).3 Results and discussion
Electrospinning of HPβCD
Previous studies on CDs have focused only on its dilute solution behavior.50 In this study we achieved solutions with high concentrations of CD, possibly due to the chemical modification of the CD molecule. The high concentrations resulted in highly viscous solutions. The relatively high viscosity led us to attempt to electrospin these solutions. Representative scanning electron microscopy (SEM) images illustrate a transition from beads to nanofibers (Fig. 2). For concentrations below 50 wt%, electrospraying occurs as only spherical beads are formed (Fig. 2a). However, at 60 wt% a mix of nanofibers and beads can be seen (Fig. 2b). At 65 wt% bead defects are absent and only fibers with an average size of 933 ± 780 nm are formed (Fig. 2c), though these fibers show variability in diameter as indicated by the large standard deviation. Increasing HPβCD concentration to 70 wt% improves fiber uniformity as the fiber size is 990 ± 120 with a much lower standard deviation, as observed in Fig. 2d. Increasing the concentration of HPβCD beyond 70 wt% led to solutions that could not be electrospun. These results indicate that polymer-free electrospun fibers can be achieved with small molecules, such as CD. Further, the ability to electrospin at high concentrations creates the opportunity for relatively high fiber throughput (by mass), overcoming the low-throughput limitation of most electrospun polymer systems. Interestingly, we generated fibers at concentrations approximately half of the 160% w/v previously reported by Uyar et al.48 as 70 wt% had a density of 1.2 g mol−1 or a 84% w/v. We speculate that the difference in concentrations may be due to a difference in sources of cyclodextrin. Working at such a different concentration range may be a result of a different aggregation mechanism than previously reported. | ||
| Fig. 2 SEM micrographs of electrospun HPβCD solutions of a) 50 wt%, b) 60 wt%, c) 65 wt%, and d) 70 wt%. | ||
Examining the fibers using high resolution TEM (Fig. 3), confirmed that submicron fibers of HPβCD were electrospun from 65 wt% aqueous solutions. The fiber surface appears rough but shows a patterned morphology at the higher magnification (Fig. 3b), possibly indicating the presence of a self-assembled structure. The dimensions of the individual features seen in Fig. 3b are similar to the dimensions of individual CD molecules. No electron diffraction pattern was obtained from the samples indicating that the CD fibers are amorphous as expected.
 | ||
Fig. 3 TEM micrographs showing fibers electrospun from a 65 wt% solution at two magnifications a) 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× and b) 400000×. 000× and b) 400000×. | ||
Solution properties
Based on these results, we investigated the solution properties that are known to affect electrospinning, specifically conductivity, surface tension, and viscosity. The conductivity for the solutions is shown in Fig. 4a. At low concentrations, the conductivity does not appear to vary with concentration; however, above 30 wt%, conductivity is inversely proportional with concentration, and thus unlikely to explain the electrospinnablity of highly concentrated CD solutions. This may be attributed to a reduction in the mobility of the bulk water. Similarly, Foster et al. reported a reduction in transport properties (including conductivity) in polymer solutions containing PEO which deviated from ideal due to the hydration of PEO and a decrease in bulk water.51 While surface tension was affected by concentration for concentrations below 20 wt%, at higher concentrations the surface tension does not appear to be affected by concentration. The surface tension of the concentrated cyclodextrin solutions were ∼60 dyne cm−1. Since we electrospin highly concentrated solutions, in the range where surface tension is unaffected, we do not discuss its effect. The viscosity of HPβCD solutions (10–80 wt%) shows a nearly exponential rise with increasing HPβCD concentration (Fig. 4b), which may be due to the formation of aggregates. Our attempts at evaluating the viscoelasticity of these solutions indicated an extremely short relaxation time as indicated by the lack of crossover of G′ and G′′ in the dynamic rheology experiment (ESI, Fig. 2 left). In fact, the system shows characteristic of a Netwonian fluid as indicated by the complex viscosity being independent of frequency (ESI, Fig. S2 right). The 45 wt% HPβCD shows an increase in complex viscosity at high frequency and this is likely an artifact of the inertia of the instrument since the sample has such a low viscosity. Based on this result, there is no indication the elasticity of the sample facilitates fiber formation.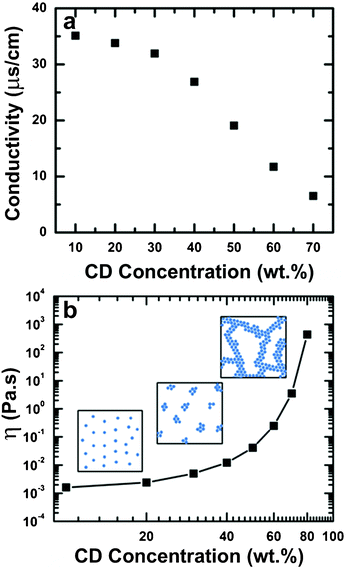 | ||
| Fig. 4 (a) Effect of HPβCD concentration on the conductivity of aqueous solutions. (b) Zero-shear viscosity for HPβCD solutions of interest, the increase in viscosity. The cartoons (not drawn to scale) show how the networks may grow larger with increasing HPβCD to the point where they have enough cohesion above 65 wt% to stretch into fibers. | ||
We used thermal analysis to further explore the electrospinnability of HPβCD and the nature of any potential CD aggregates in our solutions. Utilizing DSC, we examined the amount of free and bound water in the HPβCD solutions. We assume that bound water molecules are interacting with the cyclodextrin molecules via hydrogen bonding. For CD concentrations above 50 wt% a dramatic reduction in the amount of free water is observed until almost none exists at 70 wt% HPβCD, as the water molecules are now occupied through hydrogen bonding with CD molecules (ESI, Fig. S1). This is consistent to what Hausler et al. found in their analysis of free and bound water in concentrated HPβCD solutions.50 Above 60 wt% HPβCD, a reduction to almost 0% free water was obtained, indicating a dramatic increase in hydrogen bonding which agrees well with the aforementioned viscosity data. We hypothesize that the presence of the hydrogen bonding may prevent polymer jet breakup during the electrospinning process, thus facilitating bead-free fiber formation.
Based on these results and assuming that all of the bound water from the calorimetric measurements is hydrating the HPβCD, we estimated the amount of bound water of hydration per CD in the aqueous solutions (m). Fig. 5a shows how the moles of water per mole of HPβCD vary with concentration. At the lowest concentration studied, 10 wt%, the hydration number, m = 12.6, is similar to that found by Shikata et al.49 The slight discrepancy can be attributed to the difference in the degree of substitution. With increasing concentration, the hydration number then decreases dramatically and plateaus at higher concentrations to a value of ∼3–5. This is due to increased interactions between the HPβCD molecules as the molecules participate in intermolecular hydrogen bonding with each other instead of with water molecules. This mechanism of aggregation is analogous to depletion flocculation.52,53 Additionally, based on the amount of bound water from our analysis and the size of the aggregates previously reported, as determined by dynamic light scattering,48 we calculated that each aggregate would contain in the order of 30 cyclodextrin molecules and a molecular weight on the order of 104 g mol−1. This is considerably smaller than the polymer chains typically electrospun.
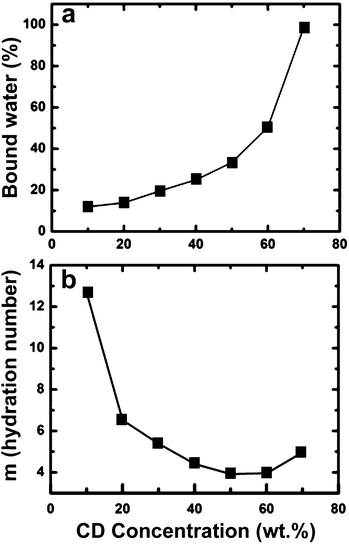 | ||
| Fig. 5 Determination of the bound water in HPβCD solutions by DSC: (a) amount of bound water present as a function of HPβCD solution concentration. (b) Hydration number as a function of HPβCD in water. | ||
This hypothesis is supported by the exponential increase of viscosity as a function of weight fraction (approximate volume fraction) in Fig. 4(b). Similar trends have been observed in concentration dispersions of weakly attracted colloidal particles.52 Additionally, the limit of the derivative of the reduced viscosity as a function of volume fractions at low concentrations approaches that predicted by the Kreiger–Dougherty equation53 (data not shown). Although we see no evidence of a network forming either in a yield stress or using dynamic rheology, we believe this may be due to the relatively weak hydrogen bonding responsible for the interaction being broken down due to shear.
An explanation for being able to electrospin CD solutions is that extensive networks of CD molecules and water form via a mechanism analogous to depletion flocculation and provide enough molecular cohesion to elongate and form fibers. Further, we observe that the onset of fiber formation can be predicted using the trend in bound water based on the hypothesis that the exclusion of water leads to an electrospinnable cyclodextrin–water network. According to DSC analysis, the hydration number falls below 6 at approximately 30 wt% (Fig. 5(b)), and we observe the onset of fiber formation to occur at 70 wt% (∼2–2.5 × Cn, where we define Cn as the onset of an electrospinnable network) when the bound water is less than 6. This result is analogous to entanglement concentration Ce for conventional electrospinning of polymers, where the polymer concentration required for electrospinning is 2–2.5 × Ce.
We probed the nature of these intermolecular interactions by examining the effect of adding a chaotropic agent to the system, specifically urea. Fig. 6 shows that by adding small amount of urea, uniform fibers (Fig. 6a) electrospun from 70 wt% HPβCD solution transition to elongated beaded fibers (Fig. 6b, 10 wt% urea), and finally to a film (Fig. 6c, 20 wt% urea). The urea reduces the interactions between the HPβCD molecules in solution causing instability and eventual breakup in the electrospinning jet. The disruption of hydrogen bonding with the addition of urea decreases the viscosity by a factor of eight as shown in Fig. 6d. This result indicates that the cyclodextrin–water network is a result of hydrogen bonding, and is consistent with previous reports of aggregation of HPβCD molecules.47,48
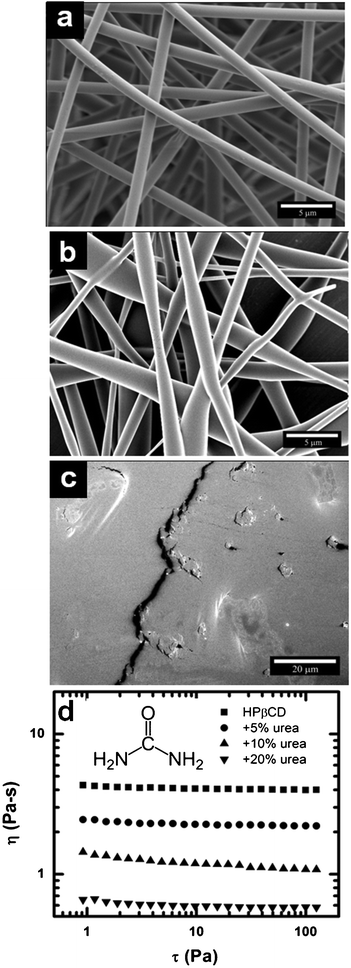 | ||
| Fig. 6 SEM micrographs of electrospun HPβCD fibers spun from solutions of a) 70 wt% HPβCD and 70 wt% HPβCD adding b) 10 wt% and c) 20 wt% urea d) Viscosity (η) versus shear stress (τ) of precursor solutions for electrospinning of both neat HPβCD and HPβCD with various amounts of urea. | ||
Other modified cyclodextrins
We have established that, with sufficient molecule cohesion, a chemically modified cyclodextrin can be electrospun to produce fibers. We extended our study to another chemically modified β-cyclodextrin, namely methyl-β-cyclodextrin produced by substituting the hydroxyl-groups present on natural β-cyclodextrin with methyl-groups, producing ether functionalities in the glucose repeating units of the CD.The corresponding SEM micrographs of electrospun submicron fibers of aqueous MβCD solutions at various concentrations are shown in Fig. 7. Fibrous material was produced starting at 60% MβCD (Fig. 7a); however, it appears to be non-continuous, having many defects and bead-like formations. As the concentration of MβCD is increased to 65% (Fig. 7b), the fibers become more regular and uniform with fewer defects, and by further increasing the concentration to 70% MβCD (Fig. 7c) continuous uniform defect free fibers comparable to the results for HPβCD (Fig. 2) are obtained. However, there appears to be a larger concentration range over which fibers can be electrospun with MβCD (60–70%) than with HPβCD (65–70%). Additionally at the same solution concentrations MβCD fibers are smaller than HPβCD fibers, despite comparable viscosities (as expected, since the hydration values were the same). For example, the fiber diameter at 70% MβCD concentration is 444 ± 59 nm whereas that of the HPβCD sample at the same concentration is ∼990 nm. The large difference in fiber size and morphology between the two species at the same concentration can be explained from differences in conductivity and surface tension (Table 1). The measured electrical conductivities were vastly different for the two CD species in solution. Methyl chloride is one of the components used in the synthesis of MβCD, which leads to a small amount (2%) of chloride ions as an impurity. These residual chloride ions serve to increase the conductivity greatly for MβCD in solution to more than 2-orders of magnitude higher than that of equivalent HPβCD aqueous solutions (Table 1). An increase in conductivity allows for a higher surface charge density to be maintained on the jet and as a result promotes improved stretching during the electrospinning process.54 In comparing this result with a previously established model,55 we find that the model overestimates the effect of conductivity. In this case the model predicts that due to the difference in conductivity the final fiber of MβCD would be 4-times smaller than HPβCD at the same concentration and process parameters, but experimentally we only see a 2-fold decrease in fiber size. However, this model was focused on linear homogeneous polymers in nonaqueous solvents, and considering the role of water in the network formation that we observe in our system, we would not necessarily expect the model to quantitatively hold for this system. Additionally, the surface tension for MβCD solutions was found to be approximately 15% lower, on average, than complementary HPβCD solutions.56 Therefore we surmise that a higher electrical conductivity combined with a lower surface tension to be responsible for both the larger electrospinnability window and the reduced fiber diameters of MβCD when compared to HPβCD.
 | ||
| Fig. 7 SEM micrographs of electrospun MβCD from aqueous solutions containing (a) 60%, (b) 65%, and (c) 70% MβCD (d) Viscosity (η) versus shear stress (τ) of precursor solutions for electrospinning of both neat MβCD and MβCD with various amounts of urea. | ||
| Concentration [wt%] | Electrical conductivity [μS cm−1] | Surface tension [dyne cm−1] |
|---|---|---|
| 60 | 91.2 | 54.2 ± 0.4 |
| 65 | 353.3 | 54.5 ± 0.4 |
| 70 | 705.0 | 54.9 ± 0.5 |
We also determined the effect of urea on this system. As aforementioned, urea is a well known hydrogen bond disruptor as demonstrated earlier with HPβCD. Again, urea proved effective in disrupting hydrogen bonds in solution reducing the viscosity (Fig. 7d) suggesting that a strong hydrogen bonding network is forming. These findings are consistent with other work.45 Thus, we have established the generality of electrospinning a hydrogen bonded CD-water network via a mechanism analogous to depletion flocculation.
4 Conclusions
We have produced oligosaccharide HPβCD submicron fibers by electrospinning highly concentrated aqueous solutions. Fibers were produced without utilizing a carrier polymer. The ability to electrospin these solutions is attributed to aggregation between HPβCD molecules at high concentrations, as evidenced from the increase in viscosity and bound water with concentration. We further examined the nature of the intermolecular interactions, and found that fiber formation is disrupted by the addition of urea, known to reduce hydrogen bonding, thus aggregation is ascribed to hydrogen bonding. We examined the generality of our results by examining other chemically modified cyclodextrins, methyl-β-cyclodextrin fibers were also produced by electrospinning concentrated aqueous solutions. The chemical modification affected concentration required to electrospin as well as fiber size at equivalent concentrations likely due to a difference in surface tension and conductivity of the solutions. These results contradict the paradigm that polymer entanglement or solution elasticity is required for electrospinning, which may lead to potential development of nanofibrous structures from a new range of materials.Acknowledgements
The authors thank Dr Orlando Rojas for his assistance with the surface tension measurements. CDS was partially supported by the STC program of the US National Science Foundation under agreement (CHE-9876674).References
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef CAS.
- A. R. Hedges, Chem. Rev., 1998, 98, 2035–2044 CrossRef CAS.
- S. Li and W. C. Purdy, Chem. Rev., 1992, 92, 1457–1470 CrossRef CAS.
- T. Loftsson, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS.
- A. E. Tonelli, Polymer, 2008, 49, 1725–1736 CrossRef CAS.
- D. M. Vriezema, M. C. Aragones, J. A. A. W. Elemans, J. J. L. M Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS.
- G. Wenz, Angew. Chem., Int. Ed. Engl., 1994, 33, 803–822 CrossRef.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS.
- R. Romi, P. Lo Nostro, E. Bocci, F. Ridi and P. Baglioni, Biotechnol. Prog., 2005, 21, 1724–1730 CrossRef CAS.
- H. J. Buschman, U. Denter, D. Knittel and E. Schollmeyer, J. Text. Inst., 1998, 89, 554–561 CrossRef.
- B. Voncina, V. Vivod and D. Jausovec, Dyes Pigm., 2007, 74, 642–646 CrossRef CAS.
- M. Tanaka, H. Matsuda, H. Sumiyoshi, H. Arima, F. Hirayama, K. Uekama and S. Tsuchiya, Chem. Pharm. Bull., 1996, 44, 416–420 CrossRef CAS.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS.
- T. Irie and K. Uekama, J. Pharm. Sci., 1997, 86, 147–162 CrossRef CAS.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS.
- A. Magnusdottir, M. Masson and T. Loftsson, J. Inclusion Phenom. Macrocyclic Chem., 2002, 44, 213–218 CrossRef CAS.
- H. Matsuda and H. Arima, Adv. Drug Delivery Rev., 1999, 36, 81–99 CrossRef CAS.
- R.A. Rajewski and V. J. J. Stella, J. Pharm. Sci., 1996, 85, 1142–1169 CrossRef CAS.
- J. Pitha, T. Irie, P. B. Sklar and J. S. Nye, Life Sci., 1988, 43, 493–502 CrossRef CAS.
- K. Uekama, Yakugaku Zasshi, 2004, 124, 909–935 CrossRef CAS.
- Z. H. Qi and A. R. Hedges, ACS Symp. Ser., 1995, 610, 231–243 CrossRef CAS.
- D. Li and Y. N. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. A. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- D. H. Reneker and I. Chun, Nanotechnology, 1996, 7, 216–223 CrossRef CAS.
- L. Li and Y. L. Hsieh, Polymer, 2005, 46, 5133–5139 CrossRef CAS.
- J. Bai, Q. Yang, M. Li, C. Zhang and Y. Li, J. Mater. Process. Technol., 2008, 208, 251–254 CrossRef CAS.
- J. Bai, Q. Yang, M. Li, S. Wang, C. Zhang and Y. Li, Mater. Chem. Phys., 2008, 111, 205–208 CrossRef CAS.
- L. F. Zhang and Y. L. Hsieh, J. Nanosci. Nanotechnol., 2008, 8, 4461–4469 CrossRef CAS.
- T. Uyar, P. Kingshott and F. Besenbacher, Angew. Chem., Int. Ed., 2008, 47, 9108–9111 CrossRef CAS.
- T. Uyar, Y. Nur, J. Hacaloglu and F. Besenbacher, Nanotechnology, 2009, 20, 125703 CrossRef.
- T. Uyar, R. Havelund, Y. Nur, J. Hacaloglu, F. Besenbacher and P. Kingshott, J. Membr. Sci., 2009, 332, 129–137 CrossRef CAS.
- T. Uyar, R. Havelund, J. Hacaloglu, X. Zhou, F. Besenbacher and P. Kingshott, Nanotechnology, 2009, 20, 125605 CrossRef.
- T. Uyar, J. Hacaloglu and F. Besenbacher, React. Funct. Polym., 2009, 69, 145–150 CrossRef CAS.
- T. Uyar and F. Besenbacher, Eur. Polym. J., 2009, 45, 1032–1037 CrossRef CAS.
- T. Uyar, A. Balan, L. Toppare and F. Besenbacher, Polymer, 2009, 50, 475–480 CrossRef CAS.
- M. G. McKee, G. L. Wilkes, R. H. Colby and T. E. Long, Macromolecules, 2004, 37, 1760–1767 CrossRef CAS.
- M. G. McKee, M. T. Hunley, J. M. Layman and T. E. Long, Macromolecules, 2006, 28, 575–583 CrossRef.
- S. L. Shenoy, W. D. Bates, H. L. Frisch and G. E. Wnek, Polymer, 2005, 46, 3372–3384 CrossRef CAS.
- S. L. Shenoy, W. D. Bates and G. Wnek, Polymer, 2005, 46, 8990–9004 CrossRef CAS.
- J. H. Yu, S. V. Fridrikh and G. C. Rutledge, Polymer, 2006, 47(13), 4789–4797 CrossRef CAS.
- S. Talwar, A. S. Krishnan, J. P. Hinestroza, B. Pourdeyhimi and S. A. Khan, Macromolecules, 2010, 43, 7650–7656 CrossRef CAS.
- Y. Dror, T. Ziv, V. Makarov, H. Wolf, A. Admon and E. Zussman, Biomacromolecules, 2008, 9, 2749–2754 CrossRef CAS.
- M. P. Cashion, X. Li, Y. Geng, M. T. Hunley and T. E. Long, Langmuir, 2010, 26, 678–683 CrossRef CAS.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 2006, 311, 353–355 CrossRef CAS.
- A. Celebioglu and T. Uyar, Chem. Commun., 2010, 46, 6903–6905 RSC.
- A. Celebioglu and T. Uyar, Langmuir, 2011, 27, 6218–6226 CrossRef CAS.
- T. Shikata, R. Takahashi and Y. Satokawa, J. Phys. Chem. B, 2007, 111, 12239–12247 CrossRef CAS.
- O. Hausler and C. C. Mullergoymann, Starch/Staerke, 1993, 45, 183–187 CrossRef.
- K. R. Foster, E. Cheever, J. B. Leonard and F. D. Blum, Biophys. J., 1984, 45, 975–984 CrossRef CAS.
- R. Buscall, J. I. McGowen and A. J. Morton-Jones, J. Rheol., 1993, 37, 621–641 CrossRef CAS.
- R. G. Larson, The structure and rheology of complex fluids, Oxford University Press, New York, 1999, 325–355 Search PubMed.
- X. H. Zong, K. Kim, D. F. Fang, S. F. Ran, B. S. Hsiao and B. Chu, Polymer, 2002, 43, 4403–4412 CrossRef CAS.
- P. K. Bhattacharjee, T. M. Schneider, M. P. Brenner, G. H. McKinley and G. C. Rutledge, J. Appl. Phys., 2010, 107, 044306 CrossRef.
- L. Leclercq, H. Bricout, S. Tilloy and E. Monflier, J. Colloid Interface Sci., 2007, 307, 481–487 CrossRef CAS.
Footnote |
| † Current address: DuPont Central Research and Development, 200 Powder Mill Road, Wilmington, DE 19880-0304 |
| This journal is © The Royal Society of Chemistry 2012 |