Synthesis of 3,5-disubstituted pyrazoles via cyclocondensation of 1,2-allenic ketones with hydrazines: application to the synthesis of 5-(5-methyl-pyrazol-3-yl)-2′-deoxycytidine†
Shenghai
Guo
*,
Jiliang
Wang
,
Dongqiang
Guo
,
Xinying
Zhang
and
Xuesen
Fan
*
School of Chemistry and Environmental Science, Henan Key Lab for Environmental Pollution Control, Henan Normal University, Xinxiang, Henan 453007, P. R. China. E-mail: shguo@htu.cn; xuesen.fan@htu.cn
First published on 16th February 2012
Abstract
An efficient procedure for the synthesis of 3,5-disubstituted pyrazoles has been achieved through cyclocondensation reaction of 1,2-allenic ketones and hydrazines under extremely mild conditions. As an application, this methodology provides a highly efficient route to 5-(5-methyl-pyrazol-3-yl)-2′-deoxycytidine.
Introduction
Pyrazoles and their derivatives, which are an important class of nitrogen-containing heterocyclic compounds, have attracted much attention due to their prevalence as scaffolds in drug discovery1 and agricultural research.2 In addition, they are attractive building blocks for the preparation of transition metal ligands3 and artificial receptors.4 Classical synthetic methods for pyrazole derivatives mostly involve the direct condensation of 1,3-dicarbonyl or α,β-unsaturated carbonyl compounds with hydrazines5 and the 1,3-dipole cycloaddition of diazo compounds onto triple bonds.6 Recently, several new approaches towards the synthesis of these compounds have been developed, including iodocyclization of α,β-alkynic hydrazones, one-pot four-step sequence consisting of Sonogashira coupling, addition-cyclocondensation, bromination, and Suzuki coupling, and Lewis acid catalyzed reaction of 3-ethoxycyclobutanones with monosubstituted hydrazines.7 Despite the many methodologies for constructing pyrazole rings summarized above, almost all of them suffer from either a lack of regiospecificity or harsh reaction conditions and multistep sequences. Therefore, a mild, efficient, and environmentally benign method for the synthesis of pyrazoles is still highly desirable. On the other hand, 1,2-allenic ketones, which are versatile and useful synthetic intermediates, can be widely used for many organic transformations, such as 1,4- or 1,2-nucleophilic addition, cycloaddition with conjugate dienes and cyclization to furans.8 We recently reported an efficient synthetic approach to highly substituted benzenes and α,β-unsaturated nitriles via tandem reaction of 1,2-allenic ketones with cyanoacetates.9 In a continuation of our studies on the reactivities of 1,2-allenic ketones, we found that the cyclocondensation of 1,2-allenic ketones with hydrazine hydrate offered an efficient synthetic strategy to substituted pyrazoles under extremely mild reaction conditions. To the best of our knowledge, there is no systematic study on this chemistry.10 Herein, we would like to report the details of our research work.Results and discussion
We began our investigation with the reaction of 1,2-allenic ketone 1a and hydrazine hydrate. We first examined the cyclocondensation of 1a with hydrazine hydrate in several organic solvents and the results are summarized in Table 1.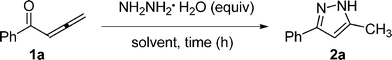
Treatment of a mixture of 1a and 1.2 equivalents of hydrazine hydrate in aprotic or protic solvent afforded the corresponding 3,5-disubstituted pyrazole 2a in 85–93% isolated yields respectively within 0.5–2 h at room temperature (Table 1, entries 1–5). It is worth noting that protic solvents are better than aprotic ones for this reaction. Reducing the amount of hydrazine hydrate from 1.2 to 1 equivalent gave 2a in the same yield (93%) using ethanol as solvent (Table 1, entries 5 and 6).
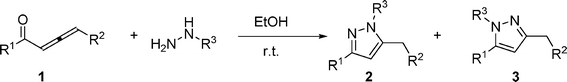
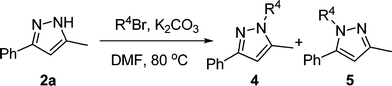
With the optimized conditions (Table 1, entry 6) in hand, we were interested in exploring the scope of 1,2-allenic ketones and hydrazines in the 3,5-disubstituted pyrazole formation reaction. The representative examples are listed in Table 2. 1-Aryl substituted allenic ketones with a wide variety of substituents on the aromatic ring reacted very well with hydrazine hydrate to afford the corresponding pyrazoles 2a–2h in good to excellent yields (Table 2, entries 1–8). The cyclocondensation reaction of 1-alkyl substituted allenic ketone 1i with hydrazine hydrate also proceeded smoothly under the same reaction conditions to generate 3,5-disubstituted pyrazole 2i in a yield of 81% (Table 2, entry 9). In addition, 1-alkyl-4-aryl or 1,4-diaryl substituted allenic ketones were all compatible with the reaction conditions to give the corresponding pyrazoles in moderate yields (Table 2, entries 10–11). Next, we examined the regioselectivity of this reaction by using substituted hydrazine as nucleophile. Unfortunately, when phenyl hydrazine was employed, two regioisomers 2l and 3l with a ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were formed in total yield of 88% (Table 2, entry 12). Formation of pyrazole regioisomers were also observed in similar studies.11 A plausible mechanism for the formation of pyrazoles 2 and 3 involving an initial 1,4-conjugate addition of hydrazine to allenic ketone followed by cyclization and aromatization was proposed in Scheme 1. Starting from 1,2-allenic ketones and monosubstituted hydrazines, pyrazoles 2 and 3 could be obtained via path a and path b, respectively. To develop an efficient approach towards 1,3,5-trisubstituted pyrazoles, we next studied the N-alkylation of pyrazole 2a with three alkyl bromides in DMF with the promotion of K2CO3. Treatment of pyrazole 2a with benzyl bromide under the standard reaction conditions afforded two regioisomers 4a and 5a with a ratio of 4.7:1 in total isolated yield of 98% (Table 3, entry 1). When propargyl and allylic bromides were employed, the N-alkylation of 2a proceeded smoothly to generate the corresponding regioisomers 4 and 5 in total yield of 91% and 70% respectively (Table 3, entries 2 and 3). These results indicated that pyrazole 4 could be regioselectively formed via the N-alkylation of 3,5-disubstituted-pyrazole 2.
:1 were formed in total yield of 88% (Table 2, entry 12). Formation of pyrazole regioisomers were also observed in similar studies.11 A plausible mechanism for the formation of pyrazoles 2 and 3 involving an initial 1,4-conjugate addition of hydrazine to allenic ketone followed by cyclization and aromatization was proposed in Scheme 1. Starting from 1,2-allenic ketones and monosubstituted hydrazines, pyrazoles 2 and 3 could be obtained via path a and path b, respectively. To develop an efficient approach towards 1,3,5-trisubstituted pyrazoles, we next studied the N-alkylation of pyrazole 2a with three alkyl bromides in DMF with the promotion of K2CO3. Treatment of pyrazole 2a with benzyl bromide under the standard reaction conditions afforded two regioisomers 4a and 5a with a ratio of 4.7:1 in total isolated yield of 98% (Table 3, entry 1). When propargyl and allylic bromides were employed, the N-alkylation of 2a proceeded smoothly to generate the corresponding regioisomers 4 and 5 in total yield of 91% and 70% respectively (Table 3, entries 2 and 3). These results indicated that pyrazole 4 could be regioselectively formed via the N-alkylation of 3,5-disubstituted-pyrazole 2.
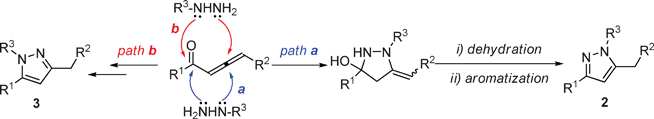 | ||
| Scheme 1 Proposed mechanism for the formation of pyrazoles 2 and 3 | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Product (ratio) | Yield (%)a |
| a Isolated yield. b Total isolated yield of 2l and 3l. | |||||
| 1 | C6H5 | H | H | 2a | 93 |
| 2 | o-FC6H4 | H | H | 2b | 86 |
| 3 | o-ClC6H4 | H | H | 2c | 92 |
| 4 | o-BrC6H4 | H | H | 2d | 96 |
| 5 | p-BrC6H | H | H | 2e | 90 |
| 6 | p-CH3C6H4 | H | H | 2f | 92 |
| 7 | p-CH3OC6H4 | H | H | 2g | 86 |
| 8 | 3,4-di-CH3OC6H3 | H | H | 2h | 93 |
| 9 | C6H5CH2 | H | H | 2i | 81 |
| 10 | CH3 | p-FC6H4 | H | 2j | 60 |
| 11 | C6H5 | C6H5 | H | 2k | 64 |
| 12 | C6H5 | H | C6H5 |
2l + 3l (1.5:1) |
88b |
C5-heteroaromatic-substituted pyrimidine nucleosides are shown to possess very important antibacterial, anti-HSV, and anticancer activities.12 The synthetic routes for these compounds mainly include: (i) [3+2] cycloaddition of 5-ethynyl pyrimidine nucleosides with organic azides or nitrile oxides;12 (ii) palladium-catalyzed cross-coupling reaction of 5-iodo pyrimidine nucleosides with heteroarylstannanes.13 However, to the best of our knowledge, efficient synthetic approaches towards 5-(5-methyl-pyrazol-3-yl) pyrimidine nucleosides have not been reported until now. To explore the bioactivities of these compounds, an efficient synthetic methodology for them is desired. As shown in Scheme 2, our methods towards 3,5-disubstituted pyrazoles from 1,2-allenic ketones have been successfully applied to the synthesis of 5-(5-methyl-pyrazol-3-yl)-2′-deoxycytidine 13. The detailed synthetic route is as follows: compound 7, which was obtained through the reaction of compound 6 with Lawesson's reagent, was methylated with MeI using Et3N as base to afford the 4-methylthio derivative 8 in good yield; The thus formed compound 8 was easily converted to 9 by using K2S2O8 as an oxidant; compound 10 prepared from the nucleophilic addition of propargylzinc bromide to aldehyde 9 was successfully oxidized to allenic ketone 11 in the presence of Jones reagent; subsequent cyclocondensation of compound 11 with hydrazine hydrate afforded compound 12 in 70% yield. The deprotection and aminolysis of 12 were achieved in a one-pot procedure by using NH3·H2O as a reactant to give product 13 in 84% yield. The structure of 13 was confirmed by 1H NMR, 13C NMR, and HRMS. Compared to the known methods toward C5-heteroaromatic-substituted pyrimidine nucleosides,12,13 the remarkable advantage of our procedure is free of expensive catalysts.
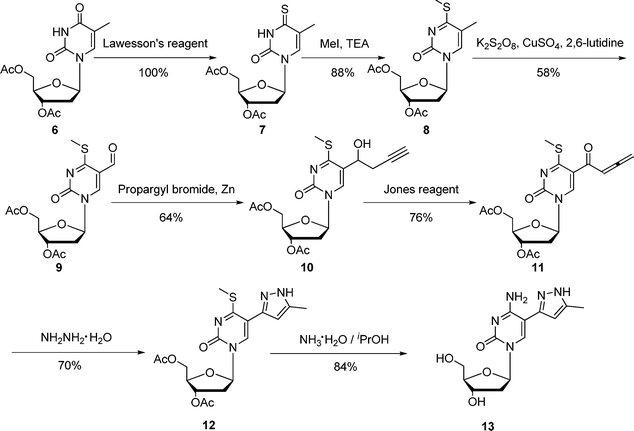 | ||
| Scheme 2 Synthesis of 5-(5-methyl-pyrazol-3-yl)-2′-deoxycytidine 13 | ||
Conclusions
In summary, we have developed a facile methodology for the synthesis of 3,5-disubstituted pyrazole derivatives through the reaction of allenic ketones and hydrazines. By using this method, 5-(5-methyl-pyrazol-3-yl)-2′-deoxycytidine was successfully prepared with good efficiency. Advantages of this method include easily obtainable starting materials, absence of expensive catalysts, and extremely mild reaction conditions. Studies to further extend the scope of this novel protocol, especially for the preparation of more pyrimidine nucleoside derivatives, are currently underway in our laboratory.Experimental
General information
THF was distilled from sodium/benzophenone. Unless noted, all commercial reagents were used without further purification. 1,2-Allenic ketones were prepared from commercially available reagents according to literature procedures.91H and 13C NMR spectra were recorded on a Bruker AC 400 MHz spectrometer at 400 and 100 MHz, respectively. All reactions were monitored by thin-layer chromatography (TLC) using silica gel plates (silica gel 60 F254 0.25 mm) and components were visualized by observation under UV light (254 and 365 nm). The characterization data of 2a7k and 2d–g7k is the same as the previously published one.General procedure for the preparation of 3,5-disubstituted pyrazole derivatives 2 and 3via cyclocondensation of 1,2-allenic ketone 1 and hydrazines
To a solution of 1,2-allenic ketone 1 (0.5 mmol) in EtOH (5 mL) was added hydrazine (0.5 mmol). The resulting solution was stirred at room temperature until the reaction was completed as monitored by TLC (ca.1 h). The solvent was evaporated in vacuo and the residue was purified by chromatography on silica-gel to afford the pyrazole products.:1–3:1) afforded the title product as red oil in 86% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.26 (s, 3H), 6.47 (d, J = 3.2 Hz, 1H), 7.09–7.14 (m, 2H), 7.22–7.25 (m, 1H), 7.81 (t, J = 7.2 Hz, 1H); 13C NMR (CDCl3, Me4Si) δ 11.64, 104.80 (d, J = 6.9 Hz, CH), 116.11 (d, J = 22.1 Hz, CH), 120.11 (d, J = 12.2 Hz, C), 124.30 (d, J = 3.0 Hz, CH), 128.28 (d, J = 3.8 Hz, CH), 129.08 (d, J = 8.3 Hz, CH), 143.41, 143.61, 159.72 (d, J = 247.7 Hz, C). HRMS (EI) calcd for C10H9FN2: 176.0750, found: 176.0743.
:1–2:1) afforded the title product as white solid in 92% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.17 (s, 3H), 6.47 (s, 1H), 7.21–7.23 (m, 2H), 7.42–7.44 (m, 1H), 7.63–7.66 (m, 1H); 13C NMR (CDCl3, Me4Si) δ 11.30, 105.75, 126.81, 128.88, 130.26, 130.61, 131.75, 132.26, 142.17, 147.14. HRMS (EI) calcd for C10H9ClN2: 192.0454, found: 192.0452.
:1–1:1) afforded the title product as yellow oil in 93% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.22 (s, 3H), 3.76 (s, 3H), 3.85 (s, 3H), 6.23 (s, 1H), 6.79 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 8.8 Hz, 1H), 7.25 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 11.58, 55.59, 55.79, 101.40, 108.85, 111.18, 118.26, 125.53, 143.25, 148.71, 148.98, 149.50. HRMS (EI) calcd for C12H14N2O2: 218.1055, found: 218.1050.
:1–2:1) afforded the title product as white solid in 81% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.27 (s, 3H), 3.99 (s, 2H), 5.83 (s, 1H), 7.24–7.33 (m, 5H); 13C NMR (CDCl3, Me4Si) δ 11.95, 33.69, 103.87, 126.32, 128.53, 128.72, 139.51, 143.52, 148.70.
:1–1:1) afforded the title product as white solid in 60% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.24 (s, 3H), 3.91 (s, 2H), 5.79 (s, 1H), 6.93–6.97 (m, 2H), 7.14–7.18 (m, 2H); 13C NMR (CDCl3, Me4Si) δ 11.79, 32.93, 103.81, 115.22 (d, J = 21.4 Hz, CH), 130.03 (d, J = 7.6 Hz, CH), 135.07 (d, J = 3.0 Hz, C), 143.15, 149.04, 161.51 (d, J = 242.3 Hz, C). HRMS (EI) calcd for C11H11FN2: 190.0906, found: 190.0909.
:1–6:1) afforded the title product as white solid in 64% isolated yield. 1H NMR (CDCl3, Me4Si) δ 3.95 (s, 2H), 6.34 (s, 1H), 7.21–7.36 (m, 8H), 7.70 (d, J = 6.8 Hz, 2H); 13C NMR (CDCl3, Me4Si) δ 33.09, 102.01, 125.77, 126.52, 127.91, 128.62, 128.74, 128.78, 132.00, 138.78, 147.69, 148.97.
:1–20:1) afforded the title product as red oil in 53% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.39 (s, 3H), 6.55 (s, 1H), 7.34–7.56 (m, 8H), 7.91–7.93 (m, 2H); 13C NMR (CDCl3, Me4Si) δ 12.58, 104.42, 124.97, 125.74, 127.61, 127.78, 128.60, 129.11, 133.41, 139.98, 140.18, 151.52.
General procedure for the preparation of 1,3,5-trisubstituted pyrazole derivatives 4 and 5via N-alkylation of pyrazole 2a with alkyl bromides
To a solution of compound 2a (0.4 mmol) in DMF (4 mL) were added alkyl bromide (2.4 mmol) and K2CO3 (3.2 mmol) at room temperature and then stirred at 80 °C for the time indicated in Table 3. Ethyl acetate and H2O were added into the reaction mixture and then extracted twice. The combined organic layer was washed with H2O and brine, dried over anhydrous Na2SO4. Ethyl acetate was removed under reduced pressure and the residue was purified by chromatography on silica-gel to afford the 1,3,5-trisubstituted pyrazoles 4 and 5.:1–10:1) afforded the title product as colorless oil in 81% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.23 (s, 3H), 5.36 (s, 2H), 6.42 (s, 1H), 7.16 (d, J = 7.6 Hz, 2H), 7.28–7.35 (m, 4H), 7.42 (t, J = 8.0 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, Me4Si) δ 11.28, 53.16, 103.28, 125.56, 126.67, 127.44, 127.54, 128.56, 128.72, 133.81, 137.12, 139.88, 150.35.
:1–10:1) afforded the title product as colorless oil in 17% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.34 (s, 3H), 5.29 (s, 2H), 6.15 (s, 1H), 7.04–7.05 (m, 2H), 7.23–7.37 (m, 8H).
:1–4:1) afforded the title product as colorless oil in 80% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.39 (overlapping, s, 4H), 4.90 (d, J = 2.4 Hz, 2H), 6.37 (s, 1H), 7.29 (t, J = 7.2 Hz, 1H), 7.38 (t, J = 7.6 Hz, 2H), 7.78 (d, J = 7.6 Hz, 2H); 13C NMR (CDCl3, Me4Si) δ 11.15, 39.23, 73.48, 77.35, 103.51, 125.55, 127.58, 128.52, 133.42, 139.83, 150.72. HRMS (EI) calcd for C13H12N2: 196.1000, found: 196.0996.
:1–4:1) afforded the title product as white solid in 11% isolated yield. 1H NMR(CDCl3, Me4Si) δ 2.32 (s, 3H), 2.42 (t, J = 2.4 Hz, 1H), 4.81 (d, J = 2.4 Hz, 2H), 6.12 (s, 1H), 7.41–7.50 (m, 5H); 13C NMR (CDCl3, Me4Si) δ 13.57, 39.16, 73.26, 78.47, 106.13, 128.65 (two peaks overlapping), 128.78, 130.29, 144.43, 148.91. HRMS (EI) calcd for C13H12N2: 196.1000, found: 196.1003.
:1–4:1) afforded the title product as colorless oil in 55% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.28 (s, 3H), 4.74 (d, J = 5.2 Hz, 2H), 5.02 (d, J = 17.6 Hz, 1H), 5.21 (d, J = 10.0 Hz, 1H), 5.97–6.05 (m, 1H), 6.36 (s, 1H), 7.29 (d, J = 7.2 Hz, 1H), 7.39 (t, J = 8.0 Hz, 2H), 7.79–7.81 (m, 2H); 13C NMR (CDCl3, Me4Si) δ 11.08, 51.88, 102.91, 116.89, 125.50, 127.37, 128.52, 133.15, 133.74, 139.78, 150.29. HRMS (EI) calcd for C13H14N2: 198.1157, found: 198.1159.
:1∼4:1) afforded the title product as colorless oil in 15% isolated yield. 1H NMR (CDCl3, Me4Si) δ 2.32 (s, 3H), 4.67 (d, J = 4.4 Hz, 2H), 5.03 (d, J = 17.6 Hz, 1H), 5.20 (d, J = 10.0 Hz, 1H), 5.95–6.05 (m, 1H), 6.10 (s, 1H), 7.37–7.44 (m, 5H); 13C NMR (CDCl3, Me4Si) δ 13.55, 51.62, 105.72, 117.18, 128.38, 128.56, 128.65, 130.86, 133.97, 144.51, 148.14. HRMS (EI) calcd for C13H14N2: 198.1157, found: 198.1157.
Procedure for the synthesis of 5-(5-methyl-pyrazol-3-yl)-2′-deoxycytidine 13
:1–1:1 as eluent to give compound 7 as yellow foam in 100% yield. 1H NMR (CDCl3, Me4Si) δ 2.12–2.21 (m, 10H), 2.52–2.58 (m, 1H), 4.28–4.42 (m, 3H), 5.22–5.24 (m, 1H), 6.25–6.29 (m, 1H), 7.35 (s, 1H), 9.65 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 17.29, 20.81, 20.89, 37.79, 63.79, 74.07, 82.49, 85.47, 119.78, 130.83, 148.00, 170.26, 170.48, 190.54. MS: m/z 343.4 [MH]+. HRMS (EI) calcd for C14H18N2O6S: 342.0886, found: 342.0873.
:1–1:4 as eluent to give compound 8 as colorless foam in 88% yield. 1H NMR (CDCl3, Me4Si) δ 2.00–2.06 (m, 10H), 2.51 (s, 3H), 2.68–2.74 (m, 1H), 4.26–4.32 (m, 3H), 5.15–5.17 (m, 1H), 6.22–6.26 (m, 1H), 7.42 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 12.84, 14.05, 20.60, 20.71, 38.43, 63.55, 74.11, 82.60, 86.60, 111.75, 135.75, 153.29, 170.00, 170.17, 178.59. MS: m/z 357.5 [MH]+. HRMS (EI) calcd for C15H20N2O6S: 356.1042, found: 356.1051.
:1–1:2 as eluent to give compound 9 as white solid in 58% yield. 1H NMR (CDCl3, Me4Si) δ 2.08 (s, 3H), 2.12 (s, 3H), 2.14–2.20 (m, 1H), 2.56 (s, 3H), 2.86–2.92 (m, 1H), 4.35–4.43 (m, 3H), 5.20–5.22 (m, 1H), 6.19–6.23 (m, 1H), 8.47 (s, 1H), 9.75 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 13.06, 20.68, 20.77, 39.00, 63.37, 73.84, 83.53, 87.88, 114.52, 148.28, 151.30, 170.27, 170.33, 177.76, 184.81. MS: m/z 371.4 [MH]+. HRMS (EI) calcd for C15H18N2O7S: 370.0835, found: 370.0840.
:1 with ethyl acetate as eluent to give compound 10 (containing two diastereoisomers) as a yellow foam in total yield of 64%. One of diastereoisomers: 1H NMR (CDCl3, Me4Si) δ 2.09–2.18 (m, 8H), 2.50–2.57 (m, 1H), 2.58 (s, 3H), 2.77–2.89 (m, 2H), 3.00 (bs, 1H), 4.32–4.40 (m, 3H), 4.92–4.97 (m, 1H), 5.23–5.25 (m, 1H), 6.32–6.35 (m, 1H), 7.97 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 12.95, 20.85, 27.03, 38.44, 63.53, 65.08, 71.77, 73.94, 80.04, 82.44, 86.41, 118.32, 136.92, 153.49, 170.48, 171.01, 175.71. MS: m/z 411.5 [MH]+. HRMS (EI) calcd for C18H22N2O7S: 410.1148, found: 410.1146.
:2 and ethyl acetate as eluent to give compound 11 as light yellow oil in 76% yield. 1H NMR (CDCl3, Me4Si) δ 1.99 (s, 3H), 2.05 (s, 3H), 2.07–2.12 (m, 1H), 2.41 (s, 3H), 2.78–2.84 (m, 1H), 4.27–4.37 (m, 3H), 5.14–5.16 (m, 1H), 5.21–5.27 (m, 2H), 6.12–6.15 (m, 2H), 8.44 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 13.98, 20.52, 20.68, 38.84, 63.37, 73.73, 79.80, 83.16, 87.40, 92.85, 113.90, 144.06, 151.09, 170.03, 170.17, 178.75, 185.11, 215.98. MS: m/z 409.3 [MH]+. HRMS (EI) calcd for C18H20N2O7S: 408.0991, found: 408.0995.
:3 ethyl acetate as eluent to give compound 12 as light yellow foam in 70% yield. 1H NMR (CDCl3, Me4Si) δ 1.87 (s, 3H), 2.02 (s, 3H), 2.09–2.17 (m, 1H), 2.25 (s, 3H), 2.43 (s, 3H), 2.65–2.69 (m, 1H), 4.22–4.24 (m, 3H), 5.15 (s, 1H), 6.23 (s, 1H), 6.27–6.30 (m, 1H), 7.91 (s, 1H); 13C NMR (CDCl3, Me4Si) δ 11.07, 13.55, 20.50, 20.83, 38.70, 63.74, 74.24, 82.78, 86.92, 104.82, 111.73, 137.59, 141.08, 143.58, 152.97, 170.41, 170.63, 177.40. MS: m/z 423.4 [MH]+. HRMS (EI) calcd for C18H22N4O6S: 422.1260, found: 422.1273.
:1–6:1 as eluent to give the final product 13 as white solid in 84% yield. 1H NMR (DMSO) δ 2.05–2.09 (m, 1H), 2.17–2.18 (m, 1H), 2.23 (s, 3H), 3.62–3.71 (m, 2H), 3.82 (s, 1H), 4.28 (s, 1H), 5.27 (s, 1H), 5.32 (s, 1H), 6.20 (t, J = 5.6 Hz, 1H), 6.29 (s, 1H), 7.86 (bs, 1H), 8.38 (bs, 1H), 8.51 (s, 1H), 12.68 (bs, 1H); 13C NMR (DMSO) δ 10.73, 41.46, 61.20, 70.19, 85.71, 87.72, 100.03, 100.35, 139.48, 139.85, 147.12, 154.29, 162.97. MS: m/z 308.6 [MH]+. HRMS (EI) calcd for C13H17N5O4: 307.1281, found: 307.1280.
Acknowledgements
We thank the Natural Science Foundation of China (20972042, 21172057) and the Doctoral Scientific Research Foundation of Henan Normal University (01036500507) for financial support.References
- (a) D. K. Walker, M. J. Ackland, G. C. James, G. J. Muirhead, D. J. Rance, P. Wastall and P. A. Wright, Xenobiotica, 1999, 29, 297 CrossRef CAS; (b) A. A. Bekhit, H. M. A. Ashour, Y. S. S. Ghany, A. E.-D. A. Bekhit and A. Baraka, Eur. J. Med. Chem., 2008, 43, 456 CrossRef CAS; (c) A. M. Farag, A. S. Mayhoub, S. E. Barakat and A. H. Bayomi, Bioorg. Med. Chem., 2008, 16, 4569 CrossRef CAS; (d) P. G. Wyatt, A. J. Woodhead, V. Berdini, J. A. Boulstridge, M. G. Carr, D. M. Cross, D. J. Davis, L. A. Devine, T. R. Early, R. E. Feltell, E. J. Lewis, R. L. McMenamin, E. F. Navarro, M. A. O'Brien, M. O'Reilly, M. Reule, G. Saxty, L. C. A. Seavers, D.-M. Smith, M. S. Squires, G. Trewartha, M. T. Walker and A. J.-A. Woolford, J. Med. Chem., 2008, 51, 4986 CrossRef CAS; (e) J. K. Yano, T. T. Denton, M. A. Cerny, X. Zhang, E. F. Johnson and J. R. Cashman, J. Med. Chem., 2006, 49, 6987 CrossRef CAS; (f) J.-Y. Shim, W. J. Welsh, E. Cartier, J. L. Edwards and A. C. Howlett, J. Med. Chem., 2002, 45, 1447 CrossRef CAS.
- (a) Y. Li, H.-Q. Zhang, J. Liu, X.-P. Yang and Z.-J. Liu, J. Agric. Food Chem., 2006, 54, 3636 CrossRef CAS; (b) H. Chen, Z. Li and Y. J. Han, J. Agric. Food Chem., 2000, 48, 5312 CrossRef CAS; (c) D. N. Gandhale, A. S. Patil, B. G. Awate and L. M. Naik, Pesticides, 1982, 16, 27 CAS.
- (a) L. Hou, W.-J. Shi, Y.-Y. Wang, H.-H. Wang, L. Cui, P.-X. Chen and Q.-Z. Shi, Inorg. Chem., 2011, 50, 261 CrossRef CAS; (b) S. Baitalik, S. Dutta, P. Biswas, U. Floerke, E. Bothe and K. Nag, Eur. J. Inorg. Chem., 2010, 570 CrossRef CAS; (c) H. Willms, W. Frank and C. Ganter, Organometallics, 2009, 28, 3049 CrossRef CAS.
- J. Chin, J. Oh, S. Y. Jon, S. H. Park, C. Walsdorff, B. Stranix, A. Ghoussoub, S. J. Lee, H. J. Chung, S.-M. Park and K. Kim, J. Am. Chem. Soc., 2002, 124, 5374 CrossRef CAS.
- (a) J. Elguero, Comprehensive Heterocyclic Chemistry, Pergamon Press, Oxford, 1984, vol. 5 Search PubMed; (b) S. Fustero, A. Simón-Fuentes and J. F. Sanz-Cervera, Org. Prep. Proced. Int., 2009, 41, 253 CrossRef CAS.
- J. Elguero, Comprehensive Heterocyclic Chemistry II, Pergamon Press, Oxford, 1996, vol. 3 Search PubMed.
- (a) For recent approaches to pyrazole derivatives, see: M. Zora, A. Kivrak and C. Yazici, J. Org. Chem., 2011, 76, 6726 CrossRef CAS; (b) M. Zora and A. Kivrak, J. Org. Chem., 2011, 76, 9379 CrossRef CAS; (c) B. Willy and T. J. J. Müller, Org. Lett., 2011, 13, 2082 CrossRef CAS; (d) G. Shan, P. Liu and Y. Rao, Org. Lett., 2011, 13, 1746 CrossRef CAS; (e) D. J. Babinski, H. R. Aguilar, R. Still and D. E. Frantz, J. Org. Chem., 2011, 76, 5915 CrossRef CAS; (f) D. Obermayer, T. N. Glasnov and C. O. Kappe, J. Org. Chem., 2011, 76, 6657 CrossRef CAS; (g) A. Tinarelli, P. Righi, G. Rosini, D. Andreotti, R. Profeta and S. Spada, Tetrahedron, 2011, 67, 612 CrossRef CAS; (h) N. Zohreh and A. Alizadeh, Tetrahedron, 2011, 67, 4595 CrossRef CAS; (i) H. Zou, H. Zhu, J. Shao, J. Wu, W. Chen, M. A. Giulianotti and Y. Yu, Tetrahedron, 2011, 67, 4887 CrossRef CAS; (j) T. Zhang, X. Gao and H. B. Wood, Tetrahedron Lett., 2011, 52, 311 CrossRef CAS; (k) J. Wen, Y. Fu, R.-Y. Zhang, J. Zhang, S.-Y. Chen and X.-Q. Yu, Tetrahedron, 2011, 67, 9618 CrossRef CAS.
- (a) S. Ma, Acc. Chem. Res., 2003, 36, 701 CrossRef CAS; (b) N. Krause, A. S. K. Hashmi, Modern Allene Chemistry, WILEY-VCH, Weinheim, 2004, vol. 2, chapter 10 Search PubMed; (c) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef; (d) H. F. Schuster and G. M. Coppola, Allenes in Organic Synthesis, Wiley, New York, 1984 Search PubMed; (e) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef; (f) L. Li and S. Ma, Chinese. J. Org. Chem., 2000, 20, 850 CAS.
- X. Zhang, X. Jia, L. Fang, N. Liu, J. Wang and X. Fan, Org. Lett., 2011, 13, 5024 CrossRef CAS.
- Few examples have been reported: M. Bertrand and M. Deline, C. R. Acad. Sc. Ser. C., 1957, 245, 2306 CAS.
- (a) J. T. Gupton, S. C. Clough, R. B. Miller, B. K. Norwood, C. R. Hickenboth, I. B. Chertudi, S. R. Cutro, S. A. Petrich, F. A. Hicks, D. R. Wilkinson and J. A. Sikorski, Tetrahedron, 2002, 58, 5467 CrossRef CAS; (b) M. Zora and M. Görmen, J. Organomet. Chem., 2007, 692, 5026 CrossRef CAS; (c) M. C. Bagley, M. C. Lubinu and C. Mason, Synlett, 2007, 704 CrossRef CAS; (d) M. Zora, A. N. Pinar, M. Odabasoğlu, O. Büyükgüngör and G. Turgut, J. Organomet. Chem., 2008, 693, 145 CrossRef CAS.
- (a) A. F. S. Ahmed, J. Chem. Res., Miniprint., 1998, 2880 Search PubMed; (b) A. Popescu, A.-B. Hoernfeldt and S. Gronowitz, Nucleos. Nucleot. Nucl., 1995, 14, 1233 CrossRef CAS; (c) P. Wigerinck, R. Snoeck, P. Claes, E. D. Clercq and P. Herdewijn, J. Med. Chem., 1991, 34, 1767 CrossRef CAS; (d) Y.-S. Lee, S. M. Park and B. H. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 1126 CrossRef CAS , and references cited therein; (e) Y.-S. Lee, S. M. Park, H. M. Kim, S.-K. Park, K. Lee, C. W. Lee and B. H. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 4688 CrossRef CAS; (f) D. W. Dodd, K. N. Swanick, J. T. Price, A. L. Brazeau, M. J. Ferguson, N. D. Jones and R. H. E. Hudson, Org. Biomol. Chem., 2010, 8, 663 RSC.
- (a) B. Froehler, US Pat., 5 645 985, 1997 Search PubMed; (b) A. J. Gutierrez, T. J. Terhorst, M. D. Matteucci and B. C. Froehler, J. Am. Chem. Soc., 1994, 116, 5540 CrossRef CAS.
- (a) E. E. Schweizer and C. S. Kim, J. Org. Chem., 1971, 36, 4033 CrossRef CAS; (b) K. Takahashi, A. Suzuki, K. Ogura and H. Iida, Heterocycles, 1986, 24, 1075 CrossRef CAS.
- J. P. Stonehouse, D. S. Chekmarev, N. V. Ivanova, S. Lang, G. Pairaudeau, N. Smith, M. J. Stocks, S. I. Sviridov and L. M. Utkina, Synlett, 2008, 100 CrossRef CAS.
- S. Peruncheralathan, T. A. Khan, H. Ila and H. Junjappa, J. Org. Chem., 2005, 70, 10030 CrossRef CAS.
- A. Alberola, L. C. Bleye, A. González-Ortega, M. L. Sádaba and M. C. Sañudo, Heterocycles, 2001, 55, 331 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2ra20274c |
| This journal is © The Royal Society of Chemistry 2012 |