Silica nanocubes with a hierarchically porous structure
Lirong
Kong
,
Xincai
Liu
*,
Xiujie
Bian
and
Ce
Wang
*
Alan G. MacDiarmid Institute, Jilin University, Changchun, P. R. China, 130012. E-mail: jfylxc@jlu.edu.cn; cwang@jlu.edu.cn; Tel: +86-431-85168292; Fax: +86-431-85168292
First published on 10th February 2012
Abstract
In this work, a simple hydrothermal method is presented for fabricating hierarchically porous silica nanocubes using a surfactant-polyelectrolyte template. The formed silica nanocubes possess Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n symmetry, which are patterned after organic template and their shape can be easily transformed from solid nanocubes to hollow cubes by controlling the composition of the surfactant-polyelectrolyte template. We have studied the fabrication conditions of silica nanocubes as a function of reaction temperature, reaction time, and the amount of reactants, and proposed a nanoparticle formation mechanism. For drug delivery, the calcined solid and hollow silica nanocubes show high loading capacities of ibuprofen, 362 mg g−1 and 509 mg g−1, respectively. These properties make the hierarchically porous silica nanocubes a promising material for drug delivery.
n symmetry, which are patterned after organic template and their shape can be easily transformed from solid nanocubes to hollow cubes by controlling the composition of the surfactant-polyelectrolyte template. We have studied the fabrication conditions of silica nanocubes as a function of reaction temperature, reaction time, and the amount of reactants, and proposed a nanoparticle formation mechanism. For drug delivery, the calcined solid and hollow silica nanocubes show high loading capacities of ibuprofen, 362 mg g−1 and 509 mg g−1, respectively. These properties make the hierarchically porous silica nanocubes a promising material for drug delivery.
1. Introduction
Due to low toxicity, large surface area and good biocompatibility, mesoporous silica has great potential for applications in catalyst loading1–3 and adsorbing,4,5 drug delivery,6–11 chromatographic separation, and membrane-based gas separation.12–14 Since the successful preparation of mesoporous SiO2 designated as M41S by the United States Mobil Corporation in 1992,15,16 more and more work has been done to enrich the family of mesoporous SiO2. For example, mesoporous silica with three dimensional pore systems, such as MCM-48,17 which provides advantages in diffusion and transport over one-dimensional channel systems,18 has also been prepared. Recently, a new surfactant-polyelectrolyte templating method has been used to prepare hierarchically porous silica microspheres with cubic Pmn symmetry by Wang et al.19 Such microspheres have an interconnected three dimensional mesopore structure. Moreover, the pores derived from the accumulation of primary silica nanoparticles further increase the surface area of these silica microspheres and provide space to accommodate other nano-sclae materials. Atluri et al. reported the preparation of amine functionalized Pmn mesoporous silica and Suteewong et al. further increased the surface density of amine groups and better controlled the sizes of particles and pores.20,21 These works further advanced the preparation technology of mesoporous silica and added new members to the family of mesoporous SiO2. However, few works have been done on controlling the shape of silica nanomaterials, as particle shapes may introduce new properties, or improve adsorptive and catalytic properties.
Controlling the shape of nanomaterials has been a challenge for chemists and materials scientists.22–25 Substantial progress has been made on the shape-controlled synthesis of noble nanoparticles (NPs) and semiconductor nanocrystals. Typical nonspherical examples include triangles, rods, cubes, and tetrapods.26–31 For example, Wang et al. reported cubic palladium NPs which have a higher catalytic activity on their exposed (100) crystal facet towards oxygen reduction reaction and a high adsorptive activity on their exposed (111) crystal facet towards sulfate ions.32 Macdonald et al. and Xia et al. reported that nanocubes, especially hollow nanocubes, could exhibit high loading capacity for drugs which may be due to their large surface area and special morphology.33,34 Based on the above report, silica with nonspherical shapes may have better performance in adsorbing in order to lower its surface energy. Furthermore, precise size control of silica NPs is also important as some of its applications are strictly size-dependant. For drug delivery, the size may affect its toxicity and the intracellular particle trafficking.35 As a result, silica NPs whose diameters are smaller than 100 nm or larger than 1000 nm are not suitable for drug delivery. However, to the best of our knowledge, precise control of size and morphology of nonspherical silica materials on nanometre scale have not been reported yet.
In this work, silica nanomaterials with a cubic structure have been successfully prepared through the surfactant-polyelectrolyte templating method. By optimizing the composition of the surfactant-polyelectrolyte template, the shape of such hierarchically porous silica material can be easily transformed from solid nanocubes to hollow nanocubes. In order to fully understand the nanocube formation mechanism, the relationship between preparation conditions (such as reaction time and temperature, etc.) and the resultant structure of the SiO2 material are studied. The SiO2 nanocubes prepared in this work exhibit high loading capacities of ibuprofen (IBU), which is a commercial antipyretic and painkilling drug.
2. Experimental section
2.1 Materials
All the reagents used in this study were analytical grade without further purification, including cetyltrimethylammonium bromide (CTAB), poly(acrylic acid) (PAA) (average molecular weight 800–1000, 30 wt% solution in water), ammonia (25%), tetraethylsiloxane (TEOS), NaH2PO4 and Na2HPO4. Water used in all experiments was distilled water.2.2 Preparation of solid silica nanocubes
In a typical experiment, CTAB (0.055 g) was completely dissolved in distilled water (40 mL) by ultrasonication, followed by adding PAA (232 μL) under vigorous stirring at room temperature. After stirring for 1 h, ammonia (192 μL) was added into the above solution, and the mixture was further stirred for 0.5 h before adding TEOS (224 μL). After stirring for another 0.5 h, the milky mixture was transferred into and sealed in a Teflon-lined stainless-steel autoclave (40 mL capacity). The autoclave was then heated and maintained at 80 °C for 48 h, and then allowed to cool down to room temperature. The obtained white product was centrifuged and washed several times with ethanol and water before being dried at 60 °C for 6 h. The organic template was removed by calcination at 550 °C for 6 h.2.3 Preparation of hollow silica nanocubes
The procedure is similar to the preparation of solid silica nanocubes except that the amount of CTAB added in distilled water (40 mL) was 0.0275 g.2.4 Drug storage and release
The drug-loaded silica nanocubes were prepared as follows: silica nanocubes (0.09 g) were dispersed in hexane–IBU solution (15 mL) with IBU concentration of 40 mg mL−1 by ultrasonication. The mixture was then sealed in a vial and stirred for 24 h. Then the IBU-adsorbed particles were separated by centrifugation for 10 min at 5000 rpm. In order to calculate IBU loading in each sample, the supernatant was removed and assessed for the content of IBU. The drug-loaded particles were washed once with hexane to remove adsorbed IBU on the exterior surface and dried at 60 °C for 12 h, which were denoted as IBU-SiO2 nanocubes and IBU-SiO2 hollow nanocubes, respectively.Before UV-vis spectroscopy measurement, calibration curves of IBU in water and hexane were obtained. We found that the IBU absorbance versus its concentration fits Lambert and Beer's law well (R = 0.999).
The in vitro release test of IBU was performed by immersing the drug-loaded nanocubes (0.05 g) in sodium phosphate buffer solution (PBS, 50 mL, pH 7.0) under mild stirring and the temperature of the releasing system was maintained at 37 °C. At selected time intervals, samples (2.0 mL) were collected from the releasing system and centrifuged to separate the supernatant and the particles. The supernatant was then analyzed by UV-vis spectroscopy to determine the amount of the released IBU. The particles were then dispersed in fresh buffer solution (2.0 mL) and put back into the releasing vessel to keep the volume constant. The adsorption peak at 263 nm was used to assess the concentration of IBU. Calculation of the corrected concentration of the released IBU is based on the following equation36
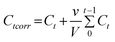 | (1) |
2.5 Characterization
Scanning electron microscopy (SEM) was performed on a SHIMADZU SSX-550 microscope. Transmission electron microscopy (TEM) experiments were performed on JEM-1200 EX (JEOL) electron microscopes with an acceleration voltage of 80 kV. Small-angle X-ray scattering (SAXS) was obtained with a Siemens D5005 diffractometer using Cu Kα radiation. Fourier transform infrared (FTIR) spectra of KBr powder-pressed pellets were collected on a BRUKER VECTOR 22 spectrometer. Thermogravimetric analysis (TGA) (PerkinElmer Pyris-1) was employed to estimate the weight percentage of the organic template. Before testing, the SiO2 material was kept in the TGA furnace at 80 °C in oxygen atmosphere for 30 min to remove any remaining residual water or solvent. The samples were measured in the range 50–750 °C at a heating rate of 10 °C min−1 in oxygen atmosphere. Ultraviolet-visible spectra (UV-vis) were collected on a Shimadzu UV-2501 PC spectrometer. Nitrogen adsorption–desorption isotherms were obtained on a TriStar 3000 nitrogen adsorption apparatus. All the samples were degassed at 200 °C for 10 h prior to Brunauer–Emmett–Teller (BET) measurements. The BET specific surface area was determined by a multipoint BET method. The desorption isotherm was used to determine the pore size distribution using the Barret–Joyner–Halenda (BJH) method. Pore volume was calculated according to the nitrogen adsorption–desorption isotherm.3. Results and discussion
3.1 Morphology investigation and formation mechanism of hierarchically porous silica nanocubes
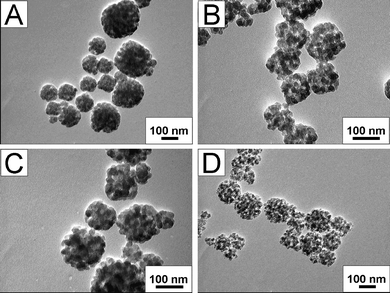
SEM and TEM images of the as-synthesized silica particles (Fig. 1) show cubic shape with sizes between 80 and 400 nm. As shown in the TEM image of SiO2 nanocubes (Fig. 1B), the nanocube is an aggregation of smaller mesoporous NPs with sizes below 10 nm. As a result, the surfaces of these nanocubes are not very smooth. The aggregated NPs form a mesoporous nanocube in which the mesopores maintain a long-range order along the [100] direction. In order to further verify that the pores were along the [100] direction, a HRTEM image of solid silica nanocubes, in which the crystal orientation was aligned along the incident electron beam could be observed with clear mesostructured fringes and cages and the Fourier diffractograms (inset in Fig. 1D), indicated that crystal orientation was along the [100] direction. This observation is similar to the results reported by Wang et al., and another similar observation is that there are a great many interstitial nanopores in the SiO2 nanocubes.19 The existence of these secondary nanopores is due to the organic template which occupies the space between the SiO2 NPs. After washing the product with water and ethanol, some of the surfactants are removed and the hierarchically mesoporous structure can be observed. Furthermore, after calcination, the hierarchical mesopores are much clearer but the morphology and mesoporous structure of these nanocubes do not change visibly (Fig. 1D).![SEM (A) and TEM (B, C) images of as-synthesized SiO2 nanocubes (A, B) and calcined SiO2 nanocubes (C), (D) HRTEM image of a calcined SiO2 nanocube and its Fourier diffractogram (inset). The HRTEM image was recorded along the direction of [100].](/image/article/2012/RA/c2ra00657j/c2ra00657j-f1.gif) | ||
| Fig. 1 SEM (A) and TEM (B, C) images of as-synthesized SiO2 nanocubes (A, B) and calcined SiO2 nanocubes (C), (D) HRTEM image of a calcined SiO2 nanocube and its Fourier diffractogram (inset). The HRTEM image was recorded along the direction of [100]. | ||
When the amount of CTAB is reduced to half, hollow SiO2 nanocubes can be obtained. From the SEM and TEM images of these hollow nanocubes in Fig. 2, it can be observed that they also have relatively uniform size (between 150 and 250 nm). As shown in the insert of Fig. 1C, during the reaction, the SiO2 NPs first aggregate into solid nanocubes or hollow nanocubes with smaller hollow space in them after 24 h and then transform to hollow nanocubes after 48 h, indicating the occurrence of Ostwald ripening.37 After calcination at 550 °C for 6 h, these nanocubes become much denser, but their particle shape and hierarchical porous structure does not change.
 | ||
| Fig. 2 SEM (A) and TEM (B, C, D) images of as-synthesized hollow SiO2 nanocubes (A, B, C) and calcined hollow SiO2 nanocubes (D). The insert TEM image in B is the magnified image of as-synthesized hollow SiO2 nanocubes and the insert TEM image in C shows the as-synthesized hollow SiO2 nanocubes prepared after a reaction time of 24 h. | ||
In order to confirm the mesostructure of these SiO2 nanocubes, SAXS has been performed. As shown in Fig. 3, three distinct peaks corresponding to (200), (210), and (211) diffractions are observed in curve a and b, indicating a Pmn mesostructure. This Pmn mesophase is patterned after the PAA-CTAB template, and then the TEOS in situ hydrolyzes in the template, leading to the formation of SiO2 nanocubes with a cubic Pmn symmetry.19,21 From the SAXS patterns in Fig. 3, it can be clearly observed that the diffraction peaks of panel b and d (calcined samples) are shifted to higher values than those of panel a and c (as-prepared samples), indicating a decrease of interplanar spacing. This shift is due to the contraction of the SiO2 nanocubes induced by further silica condensation upon calcination, and this can also explain why hollow SiO2 nanocubes are much denser after calcination (Fig. 2D). On the other hand, the diffraction peaks of hollow SiO2 nanocubes are broadened and even overlap with nearby peaks. This may be because during the Ostwald ripening process, the long-range order of the mesopores can not be kept.
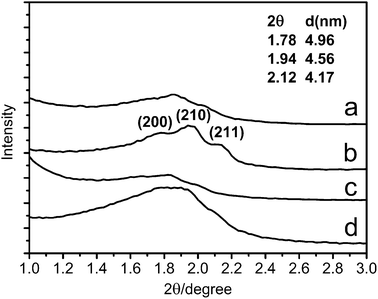 | ||
| Fig. 3 SAXS patterns of the (a) as-prepared solid SiO2 nanocubes, (b) calcined solid SiO2 nanocubes, (c) as-prepared hollow SiO2 nanocubes and (d) calcined hollow SiO2 nanocubes. | ||
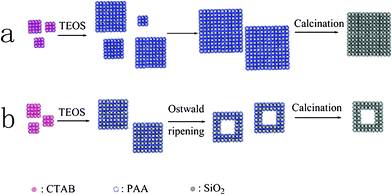 | ||
| Scheme 1 Simplified schematic representations for the fabrication of (a) solid SiO2 nanocubes and (b) hollow SiO2 nanocubes. | ||
Based on the above investigation on the morphology and structure evolvement, a tentative mechanism is proposed for the formation of these two silica nanocubes (Scheme 1). For the formation of solid silica nanocubes, the cationic surfactant CTAB and anionic polyelectrolyte PAA firstly co-organize into an ordered cubic mesophase and morphology under low concentrations by the electrostatic attraction between them. After the addition of ammonia and TEOS, the silica precursor in situ hydrolyzes in the template and thus the anisotropic silica nanocubes with a hierarchically porous structure are obtained. Along with the formation of these cubic silica NPs, they aggregate together to form larger silica nanocubes by the electrostatic interactions between PAA and CTAB, and reach maximal and relatively uniform sizes after 48 h reaction. When the amount of CTAB is reduced to half, the aggregation of silica NPs quickly terminates after 24 h reaction because of the weaker electrostatic interaction between PAA and CTAB, and this is caused by a lower amount of PAA and CTAB coated on the silica matrix. After the formation of solid silica nanocubes, as the inner small silica NPs have higher surface energy than the outer large silica NPs, they gradually dissolve and the outer silica NPs grow according to the Ostwald ripening process. However, the Ostwald ripening process could not be observed in route (a) even when we prolonged the reaction time to 96 h. This is because a large amount of PAA and CTAB coated on small silica NPs can stabilize them.
In order to further understand the formation mechanism and find the best conditions for the preparation of silica nanocubes, the effects of the reactants concentrations, reaction time, and temperature on the final morphology of the product have been carefully investigated. As shown in Fig. 4A, when the amount of CTAB is doubled, the aggregation of the SiO2 NPs is partially suppressed and more isolated SiO2 NPs are observed in the final product. Even some SiO2 NPs aggregate to form cube-like particles, the pore space between them is much larger than those prepared under normal conditions (Fig. 1B). This may be because more CTAB coating on the SiO2 NPs adsorbs more PAA through electrostatic interactions. Along with the hydrolyzation of TEOS, the PAA-CTAB composite works as a dynamic template and CTAB takes part in making the mesopores in the silica matrix while PAA serves as the template for the secondary interstitial pores inside the SiO2 nanocubes19 As a result, more PAA adsorbs on SiO2 NPs and can enlarge the space of secondary interstitial pores and stabilize the isolated SiO2 NPs. In our experiments, it is found that SiO2 materials with cubic morphology and relatively uniform sizes can not be easily obtained. As shown in Fig. 4B, it can be observed that when the concentrations of all the reactants are doubled, SiO2 materials exhibit an irregular spherical morphology. This may be due to the fast aggregation of silica NPs. Contrarily, when their concentrations are halved, the concentration of the mixed solution becomes non-uniform and thus the uniformity of silica nanocubes is affected. In detail, silica NPs aggregate into large spheres at the sites where their density is high and aggregate into smaller nanocubes at the sites where their density is low. Finally, the product is mixed with silica materials of different morphologies and sizes (Fig. 1C).
 | ||
| Fig. 4 TEM images of SiO2 nanocubes: (A) using double amount of CTAB; (B) using double amount of all the reactants (PAA, CTAB, ammonia and TEOS); (C) using half amount of all the reactants. | ||
The concentration of ammonia or TEOS has been also tuned in order to control the size and morphology of silica nanocubes. However, when catalyzed by twice the amount of ammonia, the cubic morphology of silica material become irregular (Fig. 5A) and this can be explained as follows: with excess ammonia, the hydrolyzation of TEOS is accelerated and thus more silica NPs are quickly formed. Then excess silica NPs aggregate at a faster rate, thus the mild and ordered self-assembly of silica NPs into nanocubes can not proceed normally. However, when the added amount of ammonia was halved, silica materials of different sizes but with a well cubic morphology were obtained. Moreover, there were still some isolated silica NPs in the product (Fig. 5B). According to the above analysis on the morphology formation mechanism, the reason for such a morphology can be easily understood, which is in contrary to that for the morphology in Fig. 5A. In detail, less ammonia would cause the deceleration of the hydrolyzation of TEOS, and thus the formation rate and the amount of silica NPs both decreased. Accordingly, the mild aggregation resulted in regular nanocubes. On the other hand, the average amount of PAA coated on each silica nanoparticle was increased as fewer silica NPs existed in the reaction system. As a result, more isolated silica NPs stabilized by PAA appeared and the secondary interstitial pores in aggregated nanocubes became larger which was similar to that in Fig. 4A. The effect of the concentration of TEOS to the morphology of the product was similar to that of ammonia and can be explained in similar manner. However, it should be mentioned that when the used amount of TEOS was halved, a large amount of isolated silica NPs were observed , indicating that the amount of TEOS affected the formation rate and the amount of silica NPs more than that of ammonia.
 | ||
| Fig. 5 TEM images of SiO2 nanocubes: (A) using double amount of ammonia; (B) using half amount of ammonia; (C) using double amount of TEOS; (D) using half amount of TEOS. | ||
Finally, the reaction temperature and time were tuned to see their influence to the morphology of the product. As reported previously, increasing reaction temperature would facilitate the interaction between silica precursors and CTAB, and thus silica oligomers would replace more PAA chains from the complex micelles. As a result, more disassociated PAA chains were released which filled the space between silica NPs, which resulted in larger secondary pores.19 The obtained products, which had smaller secondary pores at 40 °C and larger secondary pores at 120 °C, were in well accordance with the theory. Another point needed to be mentioned was that in Fig. 6A and 6B, silica particles prepared at 40 °C exhibit irregular shape with different sizes but those prepared at 120 °C exhibit better size uniformity and cubic morphology. This could be ascribed to the moving rate of the hydrolyzed silica NPs which is mainly affected by the temperature as the reaction proceeds without stirring. Affected by the gravity of formed silica NPs, the deposition process results in the concentration gradient. At low temperature, the silica NPs moved very slowly and their slow movement could not counteract the gravitational sedimentation. As a result, an irregular morphology appeared which was similar to that in Fig. 3C and can be explained in the same way. When the reaction temperature was increased, the gravitational sedimentation affected very little the concentration uniformity of the mixture and thus their morphology was much better. The effect of the reaction time was also studied (Fig. 6). After reacting for 24 h, incomplete growth and aggregation of silica NPs resulted in not very regular nanocubes and the sizes were also not uniform. However, after 96 h reaction, further condensation of silica precursor resulted in more silica oligomers which replaced more PAA. Thus the secondary pores became larger and the further aggregation of the silica NPs caused the appearance of spherical silica particles.
 | ||
| Fig. 6 TEM images of SiO2 nanocubes: (A) T = 40 °C; (B) T = 120 °C; (C) t = 24 h; (D) t = 96 h. | ||
3.2 Structural characterization and drug release properties of hierarchical porous silica nanocubes.
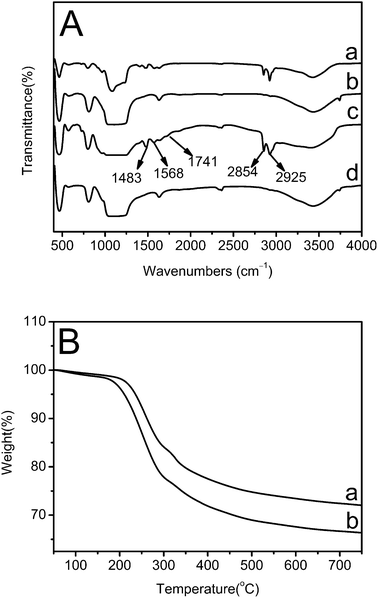
In order to determine the existing organic substances and their content in the as-prepared silica nanocubes, FTIR and TGA have been performed (Fig. 7). As shown in Fig. 7A, the spectra of as-prepared solid SiO2 nanocubes (curve a) and as-prepared hollow SiO2 nanocubes (curve c) exhibit similar peaks at 2925 cm−1, 2854 cm−1 (the C–H stretching vibrations of –CH2 in CTAB), 1741 cm−1 (the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration in PAA), 1568 and 1483 cm−1 (the C–H bending vibration in CTAB), which do not appear in the spectra of calcined silica nanocubes. The FTIR spectra indicate that the two organic substances, CTAB and PAA, are both contained in the as-prepared samples and are completely removed by calcination. As shown in Fig. 7B, during the calcination, the organic templates in the two samples both begin to decompose at around 200 °C. According to TGA results, the as-prepared hollow silica nanocubes contain 27.9% (weight percentage) of organic template while solid silica nanocubes contain 33.5% of the template.
O stretching vibration in PAA), 1568 and 1483 cm−1 (the C–H bending vibration in CTAB), which do not appear in the spectra of calcined silica nanocubes. The FTIR spectra indicate that the two organic substances, CTAB and PAA, are both contained in the as-prepared samples and are completely removed by calcination. As shown in Fig. 7B, during the calcination, the organic templates in the two samples both begin to decompose at around 200 °C. According to TGA results, the as-prepared hollow silica nanocubes contain 27.9% (weight percentage) of organic template while solid silica nanocubes contain 33.5% of the template.
 | ||
| Fig. 7 (A) FTIR spectra of (a) as-prepared solid SiO2 nanocubes, (b) calcined solid SiO2 nanocubes, (c) as-prepared hollow SiO2 nanocubes and (d) calcined hollow SiO2 nanocubes; (B) TGA curves of (a) as-prepared hollow SiO2 nanocubes and (b) as-prepared solid SiO2 nanocubes. | ||
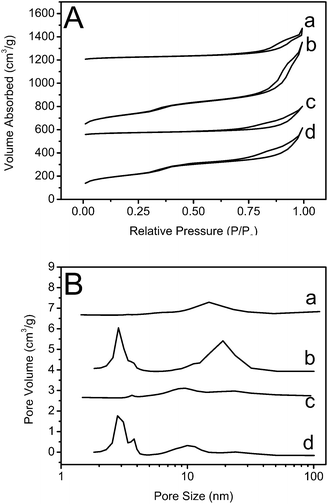
In order to investigate the hierarchical porous structures of the as-prepared and calcined silica nanocubes, nitrogen adsorption–desorption isotherms and pore-size distribution analysis have been performed (Fig. 8). As shown in Fig. 8A, according to BDDT classification,38 the four samples all exhibit type IV isotherms with H3 hysteresis loop at a relative high pressure which are due to the incomplete desorption of N2 from slitlike mesopores (2–50 nm). The calculated BET surface areas of as-prepared and calcined solid silica nanocubes are 89.90 m2 g−1 and 649.25 m2 g−1, respectively; while those of as-prepared and calcined hollow silica nanocubes are 100.10 m2 g−1 and 568.09 m2 g−1, respectively, indicating that most of the effective surface area is coated with organic substances before calcination. Moreover, the surface area of hollow silica nanocubes is higher than that of solid ones before calcination but lower after calcination. According to the TGA results, this may be because there are fewer organic substances in hollow nanocubes than those in solid ones. As a result, after removing the organic template, the surface area of hollow silica nanocubes increase less than that of solid silica nanocubes. The total mesopore volumes of solid and hollow silica nanocubes were 1.28 cm3 g−1 and 0.91 cm3 g−1 respectively, which also confirmed that different amounts of template were contained in the two samples.
 | ||
| Fig. 8 (A) Nitrogen adsorption–desorption isotherm and (B) pore-size distribution of (a) as-prepared solid SiO2 nanocubes, (b) calcined solid SiO2 nanocubes, (c) as-prepared hollow SiO2 nanocubes and (d) calcined hollow SiO2 nanocubes. | ||
From the pore-size distribution curves, a more direct result about the pore structure of the four samples can be observed (Fig. 8B). The as-prepared solid silica nanocubes only exhibit a low and wide peak centered at 14.81 nm (curve a) but the calcined solid sample exhibits three high peaks centered at 2.84 nm, 3.78 nm and 19.12 nm (curve b). According to the pore-size distribution and surface area results, it can be concluded that the mesopores with smaller sizes contribute more to the surface area of silica nanocubes. As the secondary interstitial pores have a larger size, the inside organic template PAA can be partly removed by washing with distilled water and ethanol while the CTAB, restricted in smaller mesopores, can hardly be removed by washing. As a result, the as-prepared solid silica nanocubes exhibit secondary interstitial pores with an average size of 14.81 nm. However, this conclusion does not fit well for hollow silica nanocubes. As shown in Fig. 8B, the as-prepared hollow silica nanocubes exhibit three peaks centered at 3.63 nm, 9.62 nm and 23.70 nm, which correspond to not only secondary interstitial pores, but also smaller mesopores in the silica nanocubes. This might be due to the thin walls of the hollow silica nanocubes facilitating the diffusion of both CTAB and PAA. When fewer organic templates are reserved in the mesopores of the silica matrix, smaller pores are obtained. Furthermore, Ostwald ripening also caused the diminution of the pores by the growth of silica NPs. On the other hand, it can be clearly observed that the size of secondary interstitial pores in solid samples (curve a and b) increases from 14.81 nm to 19.12 nm after calcination but this effect is weak for hollow samples (curve c and d), which is also because less organic template is contained in the hollow samples.
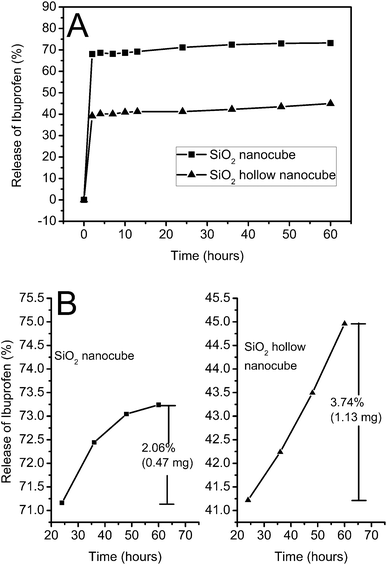
As a potential drug carrier, the drug loading and release properties of calcined silica nanocubes have been investigated (Fig. 9). In the experiments, IBU is selected as the model drug, for its short biological half-life (2 h), good pharmacological activity and the suitable molecule size (0.6–1.0 nm).39 The prepared silica nanocubes show high loading capacities of IBU and the calcination decreases the amount of functional groups on silica and thus weakens the interaction between silica nanocubes and IBU. The loading capacity of IBU is 362 mg g−1 (the weight ratio of IBU to IBU contained silica nanocubes) for calcined solid silica nanocubes, and 509 mg g−1 for calcined hollow silica nanocubes. As the surface areas are not very high when compared to previously reported results,36,40 the high loading capacities of the silica nanocubes can be attributed to their unique cubic shape, small size and the hierarchical porous structure which might facilitate the diffusion of IBU to all the pores in the silica matrix. Furthermore, the loading capacity of hollow silica nanocubes is a lot higher than that of solid ones though its surface area and total pore volume is much lower than that of the solid nanocubes. This result indicates that the hollow structure is favorable in storing small molecular drugs which can enter the large hollow space, the volume of which was not calculated in the total pore volume. From the release profile in Fig. 9, it can be observed that the drug is released much quicker in the first 2 h, which may be because a large amount of IBU adsorbed on the surface of silica nanocubes can be easily dissolved into the releasing system. As the calcined silica nanocubes have few functional groups on their surface, they have very weak interaction with IBU. As previously reported, the release process can be retarded by modifying silica materials with 3-(aminopropyl) triethoxysilane (APTS) to add amino groups on their surface.39,40 However, in order to investigate the effect of the structure on the releasing behavior of the IBU contained in silica nanocubes, no modification was made to the nanocubes. As shown in Fig. 8A, though the loading capacity of hollow silica nanocubes is a lot higher than that of the solid ones, its releasing capacity, which is 229 mg g−1, is much lower than that of the solid ones, which is 265 mg g−1. As 73.24% of the contained IBU is released from solid silica nanocubes, only 44.96% of the contained IBU is released from the hollow ones. Considering that other influencing factors were eliminated in the experiments, the lower releasing capacity of the hollow silica NPs should be ascribed to its structure. In detail, during the release of IBU from hollow silica nanocubes, smaller water molecules quickly enter the cavity and dilute the IBU inside, which slows down the releasing process because the concentration difference between the inside and the outside solution decreases. When the concentration of the outside solution is equal to that of inside solution in the cavity, the releasing process reaches equilibrium. Unless new releasing medium is added into the system to dilute it, the amount of IBU in the cavity of the hollow silica nanocubes remains constant. As a result, the IBU loaded hollow silica nanocubes release fewer drug molecules in the releasing process but it can quickly release IBU when fresh PBS is added into the releasing system. In comparison, the concentration difference does not exist in the solid silica nanocubes, and thus they quickly release IBU in the releasing process and can hardly release the drug after the releasing amount reaches a maximum value, even when the new releasing medium is added into the system. As a result, the hollow silica nanocube is a promising material for controlled release of drugs by simply controlling the concentration of drugs in the releasing system. From Fig. 9B, it can be found that during the last 36 h of the releasing process, only 0.47 mg (2.06%) of the contained IBU is released from the solid silica nanocubes and the releasing rate is a step-down when the releasing amount is 1.13 mg (3.74%) for hollow silica nanocubes and the releasing amount is in a linear relationship with time. This further confirms the conclusions suggested above.
 | ||
| Fig. 9 (A) Ibuprofen release behavior from solid SiO2 nanocubes and hollow SiO2 nanocubes; (B) amplified comparison of ibuprofen release behavior between solid SiO2 nanocubes and hollow SiO2 nanocubes. | ||
4. Conclusions
In summary, a one-step method is proposed to prepare cubic silica particles with a hierarchical porous structure. Moreover, its structure can be easily transformed from solid nanocubes to hollow ones by simply controlling the amount of the organic template. This method provides a new approach to prepare hierarchically porous particles with cubic morphology and high adsorptive activities. In our experiments, the obtained silica materials exhibit high loading capacities for IBU, which are 362 mg g−1 and 509 mg g−1 for solid and hollow nanocubes, respectively. It is also found that the releasing behavior of hollow silica nanocubes is sensitive to the concentration of the drug in the releasing system. Thus it is a promising material for the controlled release of drugs. On the other hand, this cubic silica material with hierarchically porous structure can also be used as template for other organic and inorganic nanomaterials, including polyaniline, carbon and MnO2, etc.41–44 As a result, it is also expected that the siliceous material and its derivatives may have great potential in applications of energy conversion devices, separation and purification processes.Acknowledgements
The financial support from the National 973 project (No. 2007CB936203), the basic research fund and the graduate innovation fund of Jilin University (No. 20111018) and the National Nature Science Foundation of China (Nos. 50973038) are greatly appreciated.References
- M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, S. G. Zhang, Y. Ichihashi, D. R. Park, Y. Suzuki, K. Koyano and T. Tatsumi, Catal. Today, 1998, 44, 327–332 CrossRef CAS.
- Y. Luo and J. Lin, Microporous Mesoporous Mater., 2005, 86, 23–30 CrossRef CAS.
- Y. Sakamoto, K. Okumura, Y. Kizaki, S. Matsunaga, N. Takahashi and H. Shinjoh, J. Catal., 2006, 238, 361–368 CrossRef CAS.
- J. C. Vartuli, A. Malek, W. J. Roth, C. T. Kresge and S. B. McCullen, Microporous Mesoporous Mater., 2001, 44–45, 691–695 CrossRef CAS.
- S. B. Wang and H. T. Li, Microporous Mesoporous Mater., 2006, 97, 21–26 CrossRef CAS.
- D. R. Radu, C.-Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 CrossRef CAS.
- J. Andersson, J. Rosenholm, S. Areva and M. Lindén, Chem. Mater., 2004, 16, 4160–4167 CrossRef CAS.
- P. P. Yang, S. S. Huang, D. Y. Kong, J. Lin and H. G. Fu, Inorg. Chem., 2007, 46, 3203–3211 CrossRef CAS.
- S. L. Gai, P. P. Yang, C. X. Li, W. X. Wang, Y. L. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166–1172 CrossRef CAS.
- F. Y. Qu, G. S. Zhu, H. M. Lin, W. W. Zhang, J. Y. Sun, S. G. Li and S. L. Qiu, J. Solid State Chem., 2006, 179, 2027–2035 CrossRef CAS.
- P. P. Yang, Z. W. Quan, L. L. Lu, S. S. Huang and J. Lin, Biomaterials, 2008, 29, 692–702 CrossRef CAS.
- C. Y. Liu, L. Q. Wang, W. Z. Ren and Z. H. Rong, Microporous Mesoporous Mater., 2007, 106, 35–39 CrossRef CAS.
- D.-H. Park, N. Nishiyama, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2003, 66, 69–76 CrossRef CAS.
- Y. Sakamoto, K. Nagata, K. Yogo and K. Yamada, Microporous Mesoporous Mater., 2007, 101, 303–311 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- A. Monnier, F. Schüth, Q. Huo, D. Kumar, D. Margolese, R. S. Maxwell, G. D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B. F. Chmelka, Science, 1993, 261, 1299–1303 CAS.
- M. L. Peña, Q. Kan, A. Corma and F. Rey, Microporous Mesoporous Mater., 2001, 44–45, 9–16 Search PubMed.
- J. G. Wang, H. J. Zhou, P. C. Sun, D. T. Ding and T. H. Chen, Chem. Mater., 2010, 22, 3829–3831 CrossRef CAS.
- R. Atluri, Y. Sakamoto and A. E. Garcia-Bennett, Langmuir, 2009, 25, 3189–3195 CrossRef CAS.
- T. Suteewong, H. Sai, R. Cohen, S. T. Wang and M. Bradbury, J. Am. Chem. Soc., 2011, 133, 172–175 CrossRef CAS.
- Z. L. Wang, Adv. Mater., 1998, 10, 13–30 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- A. R. Tao, S. Habas and P. D. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- Y. W. Jun, J. S. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS.
- X. G. Peng, L. Manna, W. D. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS.
- L. Manna, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 2000, 122, 12700–12706 CrossRef CAS.
- L. S. Li, J. T. Hu, W. D. Yang and A. P. Alivisatos, Nano Lett., 2001, 1, 349–351 CrossRef CAS.
- Y. W. Jun, S. M. Lee, N. J. Kang and J. Cheon, J. Am. Chem. Soc., 2001, 123, 5150–5151 CrossRef CAS.
- L. Li, Y. Yang, J. Ding and J. M. Xue, Chem. Mater., 2010, 22, 3183–3191 CrossRef CAS.
- X. A. Fan, J. G. Guan, Z. Z. Li, F. Z. Mou, G. X. Tong and W. Wang, J. Mater. Chem., 2010, 20, 1676–1682 RSC.
- C. Wang, H. Daimon, T. Onodera, T. Koda and S. H. Sun, Angew. Chem., Int. Ed., 2008, 47, 3588–3591 CrossRef CAS.
- J. E. Macdonald, M. B. Sadan, L. Houben, I. Popov and U. Banin, Nat. Mater., 2010, 9, 810–815 CrossRef CAS.
- G. D. Moon, S. W. Choi, X. Cai, W. Y. Li, E. C. Cho, U. Jeong, L. V. Wang and Y. N. Xia, J. Am. Chem. Soc., 2011, 133, 4762–4765 CrossRef CAS.
- J. M. Rosenholm, C. Sahlgren and M. Lindén, Nanoscale, 2010, 2, 1870–1883 RSC.
- Y. F. Zhu, E. Kockrick, T. Ikoma, N. Hanagata and S. Kaskel, Chem. Mater., 2009, 21, 2547–2553 CrossRef CAS.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492–3495 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- P. P. Yang, Z. W. Quan, Z. Y. Hou, C. X. Li, X. J. Kang, Z. Y. Cheng and J. Lin, Biomaterials, 2009, 30, 4786–4795 CrossRef CAS.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C. H. Shin, J. G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688–689 CrossRef CAS.
- T. H. Hsieh, K. S. Ho, X. T. Bi, Y. K. Han, Z. L. Chen, C. H. Hsu and Y. C. Chang, Eur. Polym. J., 2009, 45, 613–620 CrossRef CAS.
- G. D. Fu, J. P. Zhao, Y. M. Sun, E. T. Kang and K. G. Neoh, Macromolecules, 2007, 40, 2271–2275 CrossRef CAS.
- L. L. Hussami, R. W. Corkery and L. Kloo, Carbon, 2010, 48, 3121–3130 CrossRef CAS.
- X. H. Tang, Z. H. Liu, C. X. Zhang, Z. P. Yang and Z. L. Wang, J. Power Sources, 2009, 193, 939–943 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |