Amine-functional polysiloxanes (AFPs) as efficient polymeric organocatalyst for amino catalysis: efficient multicomponent Gewald reaction, α-allylic alkylation of aldehydes, and Knoevenagel condensation†
Zhan-Jiang
Zheng
,
Lu-Xin
Liu
,
Guang
Gao
,
Hong
Dong
,
Jian-Xiong
Jiang
,
Guo-Qiao
Lai
* and
Li-Wen
Xu
*
Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 310012, China. E-mail: liwenxu@hznu.edu.cn; licpxulw@yahoo.com; Fax: +86-571-28865135; Tel: +86-571-28867756
First published on 13th February 2012
Abstract
In this article, for the first time, we describe that commercially available and environmentally benign amine-functional polysiloxanes (AFPs) could be used as highly efficient metal-free amino catalysts in multicomponent Gewald reactions, α-allylic alkylations of aldehydes, and Knoevenagel condensations. Using catalytic amounts of AFPs, these transformations, including the Gewald reactions of ketones, sulfur, and ethyl 2-cyanoacetate, α-allylic alkylations of aldehydes and Knoevenagel condensations of aldehydes and the methylene-activated substrates may be carried out under mild conditions to give corresponding products in good yields.
1. Introduction
Amine-functional polysiloxanes (AFPs) which possess a Si–O bond in the polymeric backbone and an amino functional group in the Si–C linking side arm, are among the most important and commercially available organosilicon materials in use today.1 Similarly to general polysiloxanes, their backbones are very flexible inorganic chains, allowing easy interconversion of conformers, which are known to give interesting properties, such as heat stability, gas permeability, limited solubility with organics and water, lubricity, and unusual surface properties.2 As a result of modifications of the polysiloxanes with an amino group, they are found to be used in myriad applications in textile industry as premium-grade fabric softeners3 and in high-technology material field,4 such as membranes, adhesives, adsorption, and coatings materials. Therefore, most of applications of polysiloxanes were derived from the extraordinary flexibility and superhydrophobility of the siloxane backbone.Although the synthesis and application of various kinds of amine-functional polysiloxanes (AFPs) has attracted much attention from the viewpoints of both academic and industry fields, to the best of our knowledge, very few examples are known where the polysiloxane were used as linker or group, even as an environmentally benign supporter in organocatalyst. On the other hand, a few contributions in the literature have reported polysiloxane bearing primary or secondary amino groups that can be used as polymeric organocatalysts or ligands in homogeneous catalysis. Only Siegel et al.5 and Bergbreiter et al.6 have reported in 2004/2006 the use of polysiloxanes as soluble inorganic polymer supports for a Lewis base organocatalyst, Cinchona alkaloid, in the Michael addition of thiol and α,β-unsaturated ketones and esters, which gave low ee values (below 20% ee). Recently, we have developed a new strategy to improve the stereoselectivity in enamine catalysis by the introduction of super-hydrophobic long-chain silicone/polysiloxane as a support/functional group for a model aldol reaction. The direct aldol reaction of cyclic ketones with different aromatic aldehydes catalyzed by polysiloxane derived primary amines has been reported with high yields, good diastereoselectivity, and up to 99% ee (Scheme 1).7 With organocatalysis meeting the standards of development of green chemistry,8 we reasoned that this polysiloxane-supported amino catalysis strategy might be applicable to the transformation of aldehydes or ketone-related reactions. Herein, we report our attempts in developing commercially available amine-functional polysiloxane promoted organic transformations, in which the scope and limitation of the amine-functional polysiloxanes used as environmentally benign polymeric organocatalyst in corresponding reactions were described for the first time, such as the one-pot multicomponent Gewald reaction, allylic alkylation of aldehydes and Knoevenagel condensation. In this study, these new approaches of AFPs catalysis combine the operational simplicity of a catalytic procedure with the convenience of the direct use of environmentally benign amine-functional polysiloxanes and substrates for the preparation of synthetically useful organic compounds.
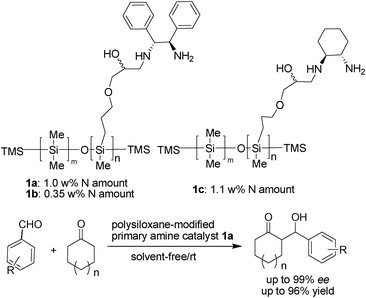 | ||
| Scheme 1 Chiral amine-functional polysiloxane-catalyzed aldol reactions. | ||
2. Results and discussion
This section is organized as follows: Different important organic transformations from simple substrates in the presence of amine-functional polysiloxanes (1d and 1e, Scheme 2) are reported through amine-functional polysiloxane-catalyzed Gewald reactions of ketones, sulfur, and ethyl 2-cyanoacetate, amine-functional polysiloxane-catalyzed α-allylic alkylations of aldehydes, and amine-functional polysiloxane-catalyzed Knoevenagel condensations.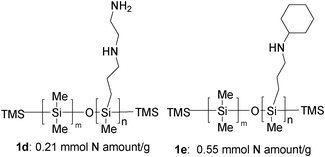 | ||
| Scheme 2 Commercially available amine-functional polysiloxanes (1d and 1e) used as polymeric organocatalysts or ligands in catalysis. | ||
2.1 Amine-functional polysiloxane-catalyzed Gewald reactions
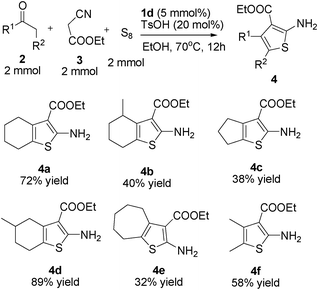
It is well known that the Gewald reaction is of considerable pharmacological importance and is a convenient procedure to synthesise substituted 2-aminothiophenes via a multicomponent condensation between sulfur, an activated methylene carbonyl compound, and α-cyanoester (Scheme 3).9 Furthermore, the thiophene scaffold is required for a broad range of important compounds including dyes, pharmaceuticals, and agrochemicals.10 Early works on Gewald reactions involving organic base-promoted multicomponent condensations have led to the development of several efficient reaction systems that achieve high levels of conversion.9 However, major problems are associated with the low reactivity of ketones, high catalyst loading (50–100 mol%), and complicated procedures. To overcome the low reactivity of the substrate, such as ketones, and to reduce the amount of organic base catalyst, it is necessary to explore a simpler, more efficient and catalytic method for the Gewald synthesis of substituted 2-aminothiophenes. | ||
| Scheme 3 Classic Gewald synthesis of substituted 2-aminothiophenes. | ||
During the previous studies on the asymmetric amino catalysis, we have found the primary amines were highly efficient catalysts in the transformation of ketones.11 Thus on the basis of previous findings, we used the primary amines and primary amine-functional polysiloxanes as catalysts for the reference Gewald reaction of cyclohexanone, sulfur, and ethyl 2-cyanoacetate. As an initial study, we noticed that the level of conversion was low when ethane-1,2-diamine was used as the catalyst. However, the primary amine-functional polysiloxane (1d) exhibited promising and better catalytic activity in this Gewald reaction (Table 1, entry 2), and as shown in Table 1, the introduction of polysiloxane provokes an increase in the conversion under the same conditions. After optimization of the reaction conditions, 20 mol% of TsOH in an EtOH solution in the presence of 5 mol% of the amine-functional polysiloxane 1d, produced the substituted 2-aminothiophene in 72% isolated yield (Table 1, entry 9). These results showed that significant functionality of the polysiloxane moiety in the amino catalysis was crucial to the multicomponent Gewald reaction, which was inconsistent with the previous findings in the aldol reaction of ketone to aldehyde.7
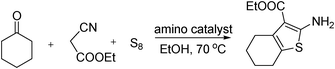
| Entry | Catalyst (x-mol N%) | Additive (x-mol%) | Time (h) | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: All reactions were performed in EtOH at 70 °C with cyclohexanone (2 mmol), sulfur (2 mmol), and ethyl 2-cyanoacetate (2 mmol) in the presence of catalyst. b Isolated yields. | ||||
| 1 | NH2(CH2)2NH2 (10) | TsOH (100) | 24 | 35 |
| 2 | 1d (5) | TsOH (100) | 12 | 58 |
| 3 | 1e (5) | TsOH (100) | 12 | 42 |
| 4 | 1d (5) | TsOH (50) | 12 | 77 |
| 5 | 1e (10) | TsOH (100) | 12 | 42 |
| 6 | 1d (5) | TFA (20) | 12 | 20 |
| 7 | 1d (5) | TfOH (20) | 12 | 20 |
| 8 | 1d (5) | AcOH (100) | 12 | 67 |
| 9 | 1d (5) | TsOH (20) | 12 | 72 |
With the optimized conditions in hand, the scope of the reaction was explored with various nonactivated ketones (Scheme 4). It was worth noting that the ring size of the cyclic ketone could influence the yield in the Gewald reaction, which was in accordance with previous reports.9e In general, good isolated yields (72–89% yield) were observed for cyclohexanone, 3-methylcyclohexanone, and 5-methylhexan-2-one, however, only moderate or low yields (32–40% yield) were achieved for cyclopentanone, 2-methylcyclohexanone, and cycloheptanone. These results showed that the amine-functional polysiloxane (1d)-catalyzed Gewald reaction was highly dependent on the nature of the ketones, which similarly to the proline-catalzyed Gewald reaction,9e for example, for 4c or 4e, only 29% or 32% yield of product was obtained respectively in the presence of L-proline. These results were poorer than those of the polysiloxane-modified primary amine catalysts. All these above experimental results showed polysiloxane-modified primary amine gave privileged reactivity in this Gewald reaction. Thus, the current study firstly demonstrated that polysiloxane was a viable functional group for the modification of primary amine-based organocatalysts, and commercially available polysiloxane functionalized primary amine-catalzyed Gewald reaction features a simple and efficient protocol for the synthesis of substituted 2-aminothiophenes.
2.2 Amine-functional polysiloxane-catalyzed α-allylic alkylation of aldehydes
Allylic alkylations are important C–C bond forming reactions in organic synthesis.12 During the last 30 years, palladium and its complexes are one of the most efficient catalysts, as a result the palladium-catalyzed allylic alkylation reaction has become a classic and mature synthetic method.13 In our preliminary communication, we have developed a very simple method for the direct, palladium-free catalytic α-allylic alkylation of aldehydes.14 The direct organocatalytic intermolecular α-allylic alkylation reaction was mediated by a simple combination of Brønsted acid and enamine catalysis which furnished α-allylic alkylated aldehydes and cyclohexanone in high yields (up to 91%) and chemoselectivities (Scheme 5). The reaction conditions are mild and environmentally friendly, the process is conducted under an atmosphere of air without the need for dried solvents, and water is the only side product of the allylic alkylation reaction. Herein, we present the investigation of the amine-functional polysiloxane catalyzed α-allylic alkylation of aldehydes.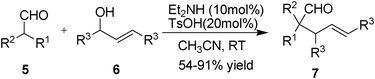 | ||
| Scheme 5 Secondary amine organocatalyzed α-allylic alkylation of aldehydes. | ||
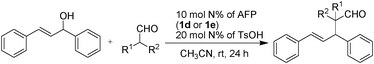
We first evaluated the catalytic activity of primary amine-functional polysiloxane 1d in the mode reaction of 1,3-diphenylprop-2-en-1-ol and isobutyraldehyde under the reported conditions,14 however, the conversion was very poor and only trace amounts of product was detected (<10% yield, Table 2, entry 1). We assumed that this low catalytic activity was due to the formation of a stable imine formed from 1d and aldehyde, and thus suppressed the intermediate for the next nucleophilic addition. When the secondary amine-functional polysiloxane (1e) was employed in this allylic alkylation of aldehyde, interestingly, high yield (86%) was achieved in this case (Table 2, entry 2). Thus we demonstrated the diversity of structures formed by this allylic alkylation of various aldehydes under the optimized reaction conditions. Except for cyclohexanecarbaldehyde and 3-methylbutanal, most of aliphatic branched or nonbranched aldehdyes gave corresponding products in good yields.
| Entry | AFP | R1 | R2 | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: All reactions were performed in CH3CN at room temperature with 1 mmol of 1,3-diphenylprop-2-en-1-ol, 2 mmol of aldehyde. b Yield of the isolated product of the corresponding product after column chromatography on silica gel, and the diastereomers could not be separated. | ||||
| 1 | 1d | Me | Me | <10 |
| 2 | 1e | Me | Me | 7a: 86 |
| 3 | 1e | H | Me | 7b: 74 |
| 4 | 1e | H | Et | 7c: 71 |
| 5 | 1e | H | n-Pr | 7d: 86 |
| 6 | 1e | H | i-Pr | 7e: 45 |
| 7 | 1e | H | CH3(CH2)7 | 7f: 89 |
| 8 | 1e | –(CH2)5– | 7g: 49 | |
| 9 | 1e | H | Ph | 7h: 83 |
| 10 | 1e | H | PhCH2 | 7i: 87 |
Along with the simple operation and good yields, the major advantage of the amine-functional polysiloxane 1e catalyst was the homogeneous reaction and heterogeneous separation. The AFP 1e exhibited very poor solubility in methanol, and the product was soluble in the methanol/CH3CN phase, therefore the methanol can be used in a liquid/gel separation with the AFP catalyst. The amine-functional polysiloxane 1e could be recovered readily from the reaction by simple centrifugal separation and could be reused at least 10 times without a decrease in the yield, and the quality of 1e was invariable during the course.
2.3 Amine-functional polysiloxane-catalyzed Knoevenagel condensations
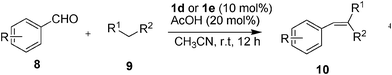
The Knoevenagel condensation is a very important and basic carbon–carbon double bond-forming reaction between a carbonyl and methylene-activated substrate, it is a widely used reaction in organic chemistry and green chemistry for the synthesis of α,β-unsaturated carbonyl compounds. Due to its widespread synthetic utility, a large number of reaction systems including some solvent-free and catalyst-free ones have been developed for this reaction.15 However, most of the currently used methods suffer from one disadvantage or another; these include the use of non-recoverable catalysts, low yields, high reaction temperatures as well as the use of toxic metals. To overcome these drawbacks, a simple, more efficient, environmentally benign method needs to be developed.On the basis of the previous studies on the Gewald reaction and the α-allylic alkylation of aldehydes, and in order to explore new applications of AFPs in organic synthesis, we then investigate the amine-functional polysiloxanes together with AcOH, in the Knoevenagel condensation of aldehydes and active methylenes. As shown in Table 3, for the most of the active methylenes tested in the condensation reactions with benzaldehyde (Table 3, entries 1–8), when AFP 1d or 1e was used, the yields of the condensation reactions were nearly quantitative. Subsequent condensations with different methylene-activated substrates and substituted aromatic aldehydes were employed gave excellent yields in most cases (Table 3, entries 10–29). Only in the case of the sterically hindered 2-bromobenzaldehyde, the condensation yield was decreased to 15% due to the steric hindrance (Table 3, entry 9). Similarly to previous reports of Knoevenagel condensations,15 ethyl cyanoacetate and malononitrile were more reactive in comparison to diethyl malonate, (Table 3, e.g., entry 5: 70% vs. entry 6: 99% and entry 7: >99% yield), due to their stronger electron-withdrawing ability.
| Entry | AFP | R | R1 | R2 | Yield (%)b |
|---|---|---|---|---|---|
| a Reaction conditions: All reactions were performed in CH3CN at room temperature with 2 mmol of aldehyde, 2.6 mmol of methylene-activated substrate, in the presence of 10 mol% of 1d or 1e. b GC yield. c Isolated yield in parentheses. | |||||
| 1 | 1d | H | CO2Et | CO2Et | 10a: 99(95)c |
| 2 | 1d | H | CN | CO2Et | 10b: 99 |
| 3 | 1d | H | CN | CN | 10c: 98 |
| 4 | 1d | H | COMe | CO2Et | 10d: 95 |
| 5 | 1e | H | CO2Et | CO2Et | 10a: 70 |
| 6 | 1e | H | CN | CO2Et | 10b: >99 |
| 7 | 1e | H | CN | CN | 10c: >99 |
| 8 | 1e | H | COMe | CO2Et | 10d: 88 |
| 9 | 1d | 2-Br | CO2Et | CO2Et | 10e: 15 |
| 10 | 1d | 4-Br | CO2Et | CO2Et | 10f: 62 |
| 11 | 1d | 2-F | CO2Et | CO2Et | 10g: 73 |
| 12 | 1d | 4-F | CO2Et | CO2Et | 10h: 94 |
| 13 | 1d | 2-Me | CO2Et | CO2Et | 10i: 42 |
| 14 | 1d | 4-Me | CO2Et | CO2Et | 10j: 98 |
| 15 | 1d | 2-OMe | CO2Et | CO2Et | 10k: 95 |
| 16 | 1d | 3-OMe | CO2Et | CO2Et | 10l: 94 |
| 17 | 1d | 4-OMe | CO2Et | CO2Et | 10m: 92 |
| 18 | 1d | 3-NO2 | CO2Et | CO2Et | 10n: 84 |
| 19 | 1d | 4-NO2 | CO2Et | CO2Et | 10o: 96 |
| 20 | 1d | 4-Cl | CN | CO2Et | 10p: 99 |
| 21 | 1d | 3-F | CN | CO2Et | 10q: 93 |
| 22 | 1d | 4-F | CN | CO2Et | 10r: 95 |
| 23 | 1d | 4-CH3 | CN | CO2Et | 10s: 92 |
| 24 | 1d | 2-OMe | CN | CO2Et | 10t: 99 |
| 25 | 1d | 3-OMe | CN | CO2Et | 10u: 99 |
| 26 | 1d | 4-OMe | CN | CO2Et | 10v: 99 |
| 27 | 1d | 2-NO2 | CN | CO2Et | 10w: 99 |
| 28 | 1d | 3-NO2 | CN | CO2Et | 10x: 99 |
| 29 | 1d | 4-NO2 | CN | CO2Et | 10y: 99 |
Furthermore, the possibility of recycling the catalyst was examined using the model reaction of benzaldehyde with malonitrile, the recycled catalyst 1d could be used at least 6 times without losing activity, and the quality of 1d decreased slightly due to poor solubility in methanol (Fig. 1). The Knoevenagel condensation product was easily extracted by methanol because the AFP 1d was not soluble in methanol and was left at the bottom of the glassware. After extraction via liquid/gel separation, the resulting AFP 1d was used directly for the next run.
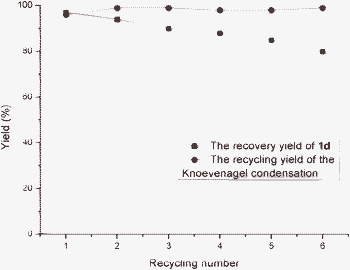 | ||
| Fig. 1 Recycling tests of the AFP 1d during the Knoevenagel condensation reaction of benzaldehyde and malonitrile. | ||
3. Conclusion
In summary, for the first time, we have found that commercially available amine-functional polysiloxanes could be used as privileged, environmentally benign, reactive and functional polymer-based organocatalysts16 in three related carbon–carbon bond-forming reactions: multicomponent Gewald reaction, α-allylic alkylation of aldehydes, and the Knoevenagel condensation. In using catalytic amounts of amine-functional polysiloxane, these transformations, including Gewald reactions of ketones, sulfur, and ethyl 2-cyanoacetate, α-allylic alkylation of aldehydes, and Knoevenagel condensations, may be carried out under mild conditions and in good to excellent yields. The superiority of the amine-functional polysiloxanes versus previous amine catalysts in terms of its stability, commercial availability, and it being non-toxic, odor-free and environmentally benign, should make it an ideal and green macromolecular amino catalyst for the preparation of various compounds via amino catalysis. Further studies will be devoted to the development of new catalytic functions and of related catalytic transformations, including enantioselective variants of the methodology described herein.4. Experimental section
4.1 General remarks
All reaction flasks and solvents were dried according to a standard method prior to use. Amine-functional polysiloxanes (AFP) 1d and 1e were purchased from Hangzhou Bald Silicone Co., Ltd. (0.21–0.55 mmol g−1 polysiloxane, Mw ∼1900). 1d, IR (cm−1): 2962, 2905, 1412, 1261, 1093, 864, 800; GPC: Mw: 2200, Mw/Mn = 2.18; 1e, IR (cm−1): 2962, 2905, 1412, 1260, 1092, 1020, 884, 800; GPC: Mw: 1900 Mw/Mn = 1.94. Gel permeation chromatographic analysis (GPC) measurements were conducted on waters GPC, Flash column chromatography was performed over silica (100–200 mesh). NMR spectra were recorded on a 400-MHz spectrometer (Avance 400). 13C-NMR spectra were obtained with broadband proton decoupling. For spectra recorded in CDCl3, unless noted, chemical shifts were recorded relative to the internal TMS (tetramethylsilane) reference signal. GC-MS was performed on TRACE DSQ. IR spectra were recorded using a FTIR apparatus (Nicolot 5700). Thin layer chromatography was performed using Silica.4.2 General procedure for amine-functional polysiloxane-catalyzed Gewald reaction
A mixture of carbonyl compound (2.0 mmol), nitrile (2 mmol), sulfur (2 mmol), amine-functional polysiloxane 1d (0.3 mmol), and EtOH (3.0 mL) in a 50 mL round bottom flask was stirred at 70 °C for 12 h. The crude product was directly purified through flash chromatography to give the desired product, substituted 2-aminothiophenes 4. The spectral data of all compounds 4 matched in all respects with reported data.84a: 1H-NMR (CDCl3, 400 MHz), δ = 5.97 (s, –NH2, 2H), 4.26 (m, –OCH2, 2H), 2.71 (t, –CH2, 2H), 2.51 (t, –CH2, 2H), 1.77 (m, –CH2, 4H), 1.33 (t, –OCH2CH3, 3H).
4b: 1H-NMR (CDCl3, 400 MHz), δ = 4.30 (m, –CH2, 2H), 3.27 (s, –MeCH, 1H), 2.49–2.52 (m, –CH2, 2H), 1.78 (m, –CH2–CH2, 4H), 1.37 (t, –CH3, 3H), 1.17 (d, –CH3, 3H).
4c: 1H-NMR (CDCl3, 400 MHz), δ = 5.82 (s, –NH2, 2H), 4.23 (t, –OCH2, 2H), 2.83 (t, –CH2, 2H), 2.71 (t, –CH2, 2H), 2.32 (m, –CH2, 2H), 1.32 (t, –OCH2CH3, 3H).
4d: 1H-NMR (CDCl3, 400 MHz), δ =5.69 (br, –NH2, 2H), 4.28 (m, –CH2, 2H), 2.93 (q, MeCH, 1H), 2.53 (s, –CH2, 2H), 2.20 (m, –H, 1H), 1.85 (m, –CH2, 4H), 1.35 (t, –CH3, 3H), 1.19 (d, –H, 1H), 1.06 (d, –CH3, 3H).
4e: 1H-NMR (CDCl3, 400 MHz), δ = 5.78 (br, –NH2, 2H), 4.28 (q, –CH2, 2H), 2.98(t, –CH2, 2H), 2.57 (t, –CH2, 2H), 1.81 (m, –CH2, 2H), 1.62 (m, –CH2–CH2, 4H), 1.35 (t, –CH3, 3H).
4f: 1H-NMR (CDCl3, 400 MHz), δ = 5.92 (s, –NH2, 2H), 4.28 (m, –OCH2, 2H), 2.17 (s, –CH3, 3H), 2.15 (s, –CH3, 3H), 1.35 (t, –OCH2CH3, 3H).
4.3 General procedure for α-allylic alkylation of aldehydes
Amine-functional polysiloxane 1d (0.1 mmol) and TsOH (0.2 mmol) was added into a solution of aldehyde (2.0 mmol) and aromatic allylic alcohol (1.0 mmol) in CH3CN (4 mL). After stirring at room temperature for 24 h, the mixture was diluted with H2O (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (Na2SO4), concentrated in vacuo, and purified by column chromatography on silica gel (EtOAc–petro ether, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) to give the pure product. All the products (7a–i) were confirmed by GC-MS, NMR, and IR.
:8) to give the pure product. All the products (7a–i) were confirmed by GC-MS, NMR, and IR.
7a (Table 2, entries 1 and 2): 1H-NMR (CDCl3, 400 MHz), δ = 1.05 (s, 3H), 1.14 (s, 3H), 3.67 (d, J = 8.8 Hz, 1H), 6.51 (m, 2H), 7.21–7.36(m, 10H), 9.62 (s, 1H). 13C-NMR (CDCl3, 100 MHz), δ = 19.66, 20.60, 49.83, 55.39, 126.37, 126.98, 127.58, 127.93, 128.40, 128.55, 129.16, 133.07, 136.96, 139.90, 206.04. MS (EI): C19H20O, m/z = 263.91. IR (cm−1), 3061, 3028, 2989, 2930, 1724, 1495, 1453, 1107, 1020.
7b (Table 2, entry 3): the diastereomers could not be separated: 1H-NMR (CDCl3, 400 MHz), δ = 1.18 (d, J = 7.2 Hz, 3H), 2.91 (m, 1H), 3.75 (t, J = 8.8 Hz, 1H), 6.32 (m, 1H), 6.49 (d, J = 16 Hz, 1H), 7.22–7.36 (m, 10H), 9.58 (d, J = 4.8 Hz, 1H). 13C-NMR (CDCl3, 100 MHz), δ = 12.24, 50.80, 50.94, 126.31, 126.95, 127.56, 127.88, 128.56, 128.91, 129.72, 131.98, 204.18. MS (EI): C18H18O, m/z = 249.89. IR (cm−1), 3060, 3027, 2988, 2934, 1724, 1599, 1494, 1453, 1153, 966.
7c (Table 2, entry 4): the diastereomers could not be separated: 1H-NMR (CDCl3, 400 MHz), δ = 0.86 (t, J = 7.6 Hz, 3H), 1.75–1.57 (m, 2H), 2.66 (m, 1H), 3.67 (t, J = 9.2 Hz, 1H), 6.22 (m, 1H), 6.41 (d, J = 15.6 Hz, 1H), 7.14–7.29 (m, 10H), 9.42 (d, J = 4.0 Hz, 1H). 13C-NMR (CDCl3, 100 MHz), δ = 19.66, 20.60, 49.83, 55.39, 126.37, 126.98, 127.58, 127.93, 128.40, 128.56, 129.16, 133.07, 136.96, 139.90, 206.04. MS (EI): C19H20O, m/z = 263.91. IR (cm−1), 3062, 3059, 3027, 2962, 2933, 2875, 1722, 1599, 1494, 1453, 1146, 967.
7d (Table 2, entry 5): the diastereomers could not be separated: 1H-NMR (CDCl3, 400 MHz), δ = 0.85 (m, 3H), 1.28 (m, 2H), 1.63 (m, 2H), 2.79 (m, 1H), 3.72 (m, 1H), 6.32 (m, 1H), 6.45 (m, 1H), 7.21–7.36 (m, 10H), 9.48 (d, J = 4.0 Hz, 0.66H), 9.64 (d, J = 4.4 Hz, 0.33H). 13C-NMR (CDCl3, 100 MHz), δ = 14.18, 20.43, 29.96, 50.27, 56.30, 126.33, 126.36, 126.96, 127.92, 128.50, 128.58, 128.67, 128.89, 128.92, 129.0, 130.23, 130.56, 131.43, 131.76, 136.89, 141.34, 204.43, 204.75. MS (EI): C20H22O, m/z = 280.82. IR (cm−1), 3061, 3027, 2957, 2931, 1724, 1494, 1147, 965.
7e (Table 2, entry 6): the diastereomers could not be separated: 1H-NMR (CDCl3, 400 MHz), δ = 1.03 (m, 6H), 1.75 (m, 0.5H), 2.18 (m, 0.5H), 2.72 (m, 1H), 3.97 (q, J1 = 9.6 Hz, J2 = 8.4 Hz, 1H), 6.25 (m, 1H), 6.47 (m, 1H), 7.21–7.36 (m, 10H), 9.57 (d, J = 4.4 Hz, 0.5H), 9.79 (d, J = 4.8 Hz, 0.5H). 13C-NMR (CDCl3, 100 MHz), δ = 17.07, 17.39, 21.61, 21.80, 27.98, 28.55, 47.81, 48.15, 61.18, 61.45, 126.30, 126.36, 126.93, 127.54, 127.97, 128.06, 128.52, 128.58, 128.69, 128.97, 130.87, 130.99, 131.11, 131.35, 136.83, 136.95, 141.44, 161.62, 205.33, 205.92. MS (EI): C20H22O, m/z = 280.80. IR (cm−1), 3060, 3026, 2960, 2872, 1720, 1494, 1452, 1144, 966.
7f (Table 2, entry 7): the diastereomers could not be separated: 1H-NMR (CDCl3, 400 MHz), δ = 0.86 (t, J = 6.8 Hz, 3H), 1.22 (m, 12H), 1.66 (m, 2H), 2.77 (m, 1H), 3.71 (q, J1 = 8.8 Hz, J2 = 8.4 Hz, 1H), 6.32 (m, 1H), 6.45 (m, 1H), 7.17–7.35 (m, 10H), 9.48 (d, J = 3.6 Hz, 0.5 H), 9.63 (d, J = 4.0 Hz, 0.5 H). 13C-NMR (CDCl3, 100 MHz), δ = 14.28, 22.19, 22.80, 27.03, 27.26, 29.28, 29.34, 29.39, 29.49, 29.57, 29.78, 31.97, 32.05, 32.09, 43.97, 50.03, 50.32, 56.53, 56.65, 126.44, 126.48, 127.02, 127.62, 128.06, 128.17, 128.60, 128.65, 128.99, 129.05, 130.40, 130.75, 131.54, 131.86, 136.96, 137.05, 141.42, 141.52, 204.23, 204.57. MS (EI): C25H32O, m/z = 348.20. IR (cm−1), 3061, 3027, 2925, 2854, 1726, 1495, 1454, 1072, 865.
7g (Table 2, entry 8): 1H-NMR (CDCl3, 400 MHz), δ = 1.21 (m, 6H), 1.52–2.08 (m, 4H), 3.36 (d, J = 9.6 Hz, 1H), 6.34 (d, J = 15.6 Hz, 1H), 6.47 (q, J = 15.2 Hz, 1H), 7.07–7.28 (m, 10H), 9.49 (s, 1H). 13C-NMR (CDCl3, 100 MHz), δ = 22.88, 23.00, 25.51, 25.70, 27.08, 30.31, 30.41, 52.98, 57.68, 126.38, 126.93, 127.51, 127.81, 128.36, 128.54, 129.11, 132.84, 137.03, 139.64, 208.32. MS (EI): C22H24O, m/z = 303.80. IR (cm−1), 3059, 3026, 2925, 2853, 1721, 1467, 1449, 1092, 964.
4.4 General procedure for the amine-functional polysiloxane-catalyzed Knoevenagel condensations
Acetic acid (20 mol%) and amine-functional polysiloxane 1d (10 mol%) was added into a solution of aldehyde (2 mmol) and a methylene-activated substrate (2.6 mmol) in CH3CN (3 mL). After stirring at room temperature for 12 h, methanol (10 mL) was added and the supernatant liquid was dried, concentrated in vacuo, and purified by column chromatography on silica gel (hexane–EtOAc, 20:1) to give the desired product.
4.5. General procedure for the recovery and recycling of the amine-functional polysiloxane 1d in the Knoevenagel condensation reactions
Acetic acid (20 mol%) and amine-functional polysiloxane 1d (10 mol%) was added to a solution of aldehyde (2 mmol) and malononitrile (2.6 mmol) in CH3CN (3 mL). After stirring at room temperature for 12 h the reaction was complete, as detected by TLC, and then methanol (10 mL) was added to the reaction solution. The product was easily extracted by methanol because the AFP 1d was not soluble in methanol and was left at the bottom of the glassware. After extraction via liquid/gel separation, the resulting AFP 1d was used directly for the next run.Acknowledgements
Financial support by the National Natural Science Founder of China (no. 20973051 and 21173064), Program for Excellent Young Teachers in Hangzhou Normal University (HNUEYT, JTAS 2011-01-014), and Zhejiang Provincial Natural Science Foundation of China (Y4090139) is appreciated. XLW is greatly indebted to Prof. Shibasaki Masakatsu, Graduate School of Pharmaceutical Sciences, The University of Tokyo, and Institute of Microbial Chemistry, Tokyo.References
- (a) S. C. Ni and P. L. Kuo, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1795 CrossRef CAS; (b) Y. Kaneko, N. Iyi, T. Matsumoto, K. Fujii, K. Kurashima and T. Fujita, J. Mater. Chem., 2003, 13, 2058 RSC; (c) M. M. Collia, M. Darder, P. Aranda and E. Ruiz-Hitzky, J. Mater. Chem., 2005, 15, 3844 RSC; (d) M. Colilla, A. J. Salinas and M. Vallet-Regi, Chem. Mater., 2006, 18, 5676 CrossRef CAS.
- (a) R. G. Jones, W. Ando and J. Chojnowski, Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications, Kluwer Academic Publishers, Dordrecht, 2000, 1–768 Search PubMed; (b) P. Izak, M. Kockerling and U. Kragl, Green Chem., 2006, 8, 947 RSC; (c) N. Benmouhoub, N. Simmonet, N. Agoudjil and T. Coradin, Green Chem., 2008, 10, 957 RSC; (d) T. Yu, K. Wakuda, D. L. Blair and R. G. Weiss, J. Phys. Chem., 2009, 113, 11546 CAS; (e) N. Shirahata, D. Hirakawa and Y. Sakka, Green Chem., 2010, 12, 2139 RSC.
- (a) M. W. Sjinner, C. Qian, S. Grigoras, D. J. Halloran and B. L. Zimmerman, Text. Res. J., 1999, 69, 935 CrossRef; (b) T. J. Kang and M. S. Kim, Text. Res. J., 2001, 71, 295 CrossRef CAS.
- (a) S. C. Ni, P. L. Kuo and J. F. Lin, Wear, 2002, 253, 862 CrossRef CAS; (b) F. Sun, S. I. Jiang and H. G. Du, J. Appl. Polym. Sci., 2008, 107, 2944 CrossRef CAS; (c) Y. L. Zub, I. V. Melnyk, M. G. White and B. Alonso, Adsorpt. Sci. Technol., 2008, 26, 119 CrossRef CAS; (d) K. Xie, L. Xu and Y. Shi, J. Fiber Bioeng. Inf., 2008, 1, 71 Search PubMed; (e) M. Li, Q. An, L. Huang, G. Yang and Q. Wang, J. Appl. Polym. Sci., 2009, 111, 2715 CrossRef CAS; (f) K. Xie, Y. Chen, A. Hou and Y. Shi, J. Dispersion Sci. Technol., 2009, 30, 1474–1480 CrossRef CAS; (g) Q. An, K. Wang and Y. Jia, Appl. Surf. Sci., 2011, 257, 4569 CrossRef CAS; (h) Y. Xu, H. Yin, H. Zheng, S. Yuan and Z. Chen, J. Appl. Polym. Sci., 2011, 119, 2326 CrossRef CAS.
- M. S. DeClue and J. S. Siegel, Org. Biomol. Chem., 2004, 2, 2287 CAS.
- M. A. Grunlan, K. R. Regan and D. E. Bergbreiter, Chem. Commun., 2006, 1715 RSC.
- L. W. Xu, Y. D. Ju, L. Li, H. Y. Qiu, J. X. Jiang and G. Q. Lai, Tetrahedron Lett., 2008, 49, 7037 CrossRef CAS.
- (a) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007, 1–536 Search PubMed; (b) B. List, Chem. Commun., 2006, 819–824 RSC , and references therein.; (c) P. J. Chua, B. tan and G. F. Zhong, Green Chem., 2009, 11, 543 RSC; (d) L. W. Xu, L. Li and Z. H. Shi, Adv. Synth. Catal., 2010, 352, 243 CrossRef CAS; (e) L. C.. Feng, Y. W. Sun, W. J. Tang, L. J. Xu, K. L. Lam, Z. Y. Zhou and A. S. C. Chan, Green Chem., 2010, 12, 949 RSC.
- (a) K. Gewald, E. Schinke and H. Böttcher, Chem. Ber., 1966, 99, 94 CrossRef CAS; (b) R. W. Sabnis, D. W. Rangnekar and N. D. Sonawane, J. Heterocycl. Chem., 1999, 36, 333 CrossRef CAS; (c) B. P. McKibben, C. H. Cartwright and A. L. Castelhano, Tetrahedron Lett., 1999, 40, 5471 CrossRef CAS; (d) M. M. Mojtahedi, M. S. Abaee, P. Mahmoodi and M. Adib, Synth. Commun., 2010, 40, 2067 CrossRef CAS; (e) T. Wang, X. G. Huang, J. Liu, B. Li, J. J. Wu, K. X. Chen, W. L. Zhu, X. Y. Xu and B. B. Zeng, Synlett., 2010, 1351 CAS.
- (a) E. Duval, A. Case, R. L. Stein and G. D. Cuny, Bioorg. Med. Chem. Lett., 2005, 15, 1885 CrossRef CAS; (b) M. Gütschow, L. Kuerschner, U. Neumann, M. Pietsch, R. Löser, N. Koglin and K. J. Eger, J. Med. Chem., 1999, 42, 5437 CrossRef; (c) H. Lutjens, A. Zickgraf, H. Figler, J. Linden, R. A. Olsson and P. J. Scammells, J. Med. Chem., 2003, 46, 1870 CrossRef; (d) D. M. Barnes, A. R. Haight, T. Hameury, M. A. McLaughlin, J. Z. Mei, J. S. J. Tedrow and D. R. Toma, Tetrahedron, 2006, 62, 11311 CrossRef CAS; (e) S. Alesi, F. Di Maria, M. Melucci, D. J. Macquarrie, R. Luque and G. Barbarella, Green Chem., 2008, 10, 517 RSC; (f) H. Li, J. Huang, L. Chen, X. Liu, T. Chen, J. Zhu, W. Lu, X. Shen, J. Li, H. Rolf and H. Jiang, J. Med. Chem., 2009, 52, 4936 CrossRef CAS; (g) J. Roger, F. Pozgan and H. Doucet, Green Chem., 2009, 11, 425 RSC; (h) T. Chonan, D. Wakasugi, D. Yamamoto, M. Yashiro, T. Oi, H. Tanaka, A. Ohoka-Suigita, I. Fusayo, H. Koretsune and A. Hiratate, Bioorg. Med. Chem., 2011, 19, 1580 CrossRef CAS.
- (a) J. Luo, H. Wang, X. Han, L. W. Han, J. Kwiatkowski, K. W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 1861 CrossRef CAS; (b) Y. F. Cai, H. M. Yang, L. Li, K. Z. Jiang, G. Q. Lai, J. X. Jiang and L. W. Xu, Eur. J. Org. Chem., 2010, 4986 CrossRef CAS; (c) H. M. Yang, L. Li, K. Z. Jiang, J. X. Jiang, G. Q. Lai and L. W. Xu, Tetrahedron, 2010, 66, 9708 CrossRef CAS; (d) H. M. Yang, Y. H. Gao, L. Li, Z. Y. Jiang, G. Q. Lai, C. G. Xia and L. W. Xu, Tetrahedron Lett., 2010, 51, 3836 CrossRef CAS; (e) Z. Y. Jiang, H. M. Yang, Y. D. Ju, L. Li, M. X. Luo, G. Q. Lai, J. X. Jiang and L. W. Xu, Molecules, 2010, 15, 2551 CrossRef CAS; (f) L. W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC; (g) J. Luo, L. W. Xu, R. A. S. Hay and Y. Lu, Org. Lett., 2009, 11, 437 CrossRef CAS; (h) L. W. Xu and Y. Lu, Org. Biomol. Chem., 2008, 6, 2047 RSC; (i) Y. D. Ju, L. W. Xu, L. Li, G. Q. Lai, H. Y. Qiu, J. X. Jiang and Y. Lu, Tetrahedron Lett., 2008, 49, 6773 CrossRef CAS.
- (a) B. M. Trost, T. R. Verhoeven, in Comprehensive Organometallic Chemistry, ed. W. Bartmann and K. B. Sharpless, Pergamon Press, Oxford, 1982, vol. 8, ch. 57, 799–938 Search PubMed; (b) D. Caine, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, pp. 1–63 Search PubMed; (c) L. Leclercq, I. Suisse, G. Nowogrocki and F. Agbossou-Niedercorn, Green Chem., 2007, 9, 1097 RSC; (d) Y. L. Liu, L. Liu, Y. L. Wang, Y. C. Han, D. Wang and Y. J. Chen, Green Chem., 2008, 10, 635 RSC.
- (a) B. M. Trost, Acc. Chem. Res., 1980, 13, 385 CrossRef CAS; (b) J. Tsuji, I. Minami and I. Shimizu, Chem. Lett., 1983, 1325 CrossRef CAS; (c) B. M. Trost and D. L. van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS.
- L. W. Xu, G. Gao, F. L. Gu, H. Sheng, L. Li, G. Q. Lai and J. X. Jiang, Adv. Synth. Catal., 2010, 352, 1441 CrossRef CAS.
- (a) A. K. Mitra, A. De and N. Karchaudhuri, Synth. Commun., 1999, 29, 2731 CrossRef CAS; (b) T. Jackson, J. H. Clark, D. J. Macquarrie and J. H. Brophy, Green Chem., 2004, 6, 193 RSC; (c) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS; (d) R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767 RSC; (e) Y. Zhang and C. G. Xia, Appl. Catal., A, 2009, 366, 141 CrossRef CAS; (f) M. Gora, B. Kozik, K. Jamrozy, M. K. Luczynski, P. Brzuzan and M. Wozny, Green Chem., 2009, 11, 863 RSC; (g) R. H. Qiu, Y. M. Qiu, S. F. Yin, X. X. Song, Z. G. Meng, X. H. Xu, X. W. Zhang, S. L. Luo, C. T. Au and W. Y. Wong, Green Chem., 2010, 12, 1767 RSC; (h) Y. F. Lai, H. Zheng, S. J. Chai, P. F. Zhang and X. Z. Chen, Green Chem., 2010, 12, 1917 RSC; (i) D. Z. Xu, Y. J. Liu, S. Shi and Y. M. Wang, Green Chem., 2010, 12, 514 RSC; (j) A. Dandia, V. Parewa, A. K. Jain and K. S. Rathore, Green Chem., 2011, 13, 2135 RSC; (k) E. Soleimani, M. M. Khodaei, N. Batooie and M. Bagbanzadeh, Green Chem., 2011, 13, 566 RSC.
- (a) K. Shiro and U. Hiroshi, Curr. Org. Chem., 2002, 6, 209 CrossRef; (b) M. Trilla, R. Plexixata, M. W. C. Man and C. Bied, Green Chem., 2009, 11, 1815 RSC; (c) A. Bancon-Caballero, G. Guillena and C. Najera, Green Chem., 2010, 12, 1599 RSC; (d) G. W. Li, J. Xiao and W. Q. Zhang, Green Chem., 2011, 13, 1828 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra00029f |
| This journal is © The Royal Society of Chemistry 2012 |