Palladium nanoshells coated with self-assembled monolayers and their catalytic properties
Jun-Hyun
Kim
*a,
Joon-Seo
Park
b,
Hae-Won
Chung
c,
Brett W.
Boote
a and
T. Randall
Lee
*c
aDepartment of Chemistry, Illinois State University, Normal, IL 61790-4160, USA. E-mail: jkim5@ilstu.edu; Fax: +1-309-438-5538; Tel: +1-309-438-2604
bDepartment of Chemistry, Eastern University, 1300 Eagle Road, St. Davids, PA 19087-3696, USA. Fax: +1-610-341-1466; Tel: +1-610-341-1485
cDepartment of Chemistry and the Texas Center for Superconductivity, University of Houston, Houston, TX 77204-5003, USA. E-mail: trlee@uh.edu; Fax: +1-281-754-4445; Tel: +1-713-743-2724
First published on 13th March 2012
Abstract
This report describes the preparation and characterization of palladium nanoshells protected with alkanethiol self-assembled monolayers (SAMs) and their application as efficient catalysts. Monodispersed silica core particles (∼100 nm in diameter) were prepared and coated with a thin layer of palladium (∼20 nm in thickness). Subsequently, the palladium nanoshells were treated with two separate alkanethiol adsorbates having different alkyl chain lengths: dodecanethiol (C12SH) and hexadecanethiol (C16SH). The optical properties, morphology, and chemical structure/composition of these nanoshells were thoroughly examined by ultraviolet-visible spectroscopy, field-emission scanning electron microscopy, transmission electron microscopy, energy-dispersive X-ray spectroscopy, X-ray photoelectron spectroscopy, and Fourier-transform infrared spectroscopy. Additional studies revealed that these SAM-coated palladium nanoshells possessed enhanced colloidal stability in nonpolar solvents and in the solid state. Further, palladium nanoshells modified with C16SH SAM coatings were employed in the Suzuki coupling of phenylboronic acid with iodobenzene in organic solvents. Notably, these SAM-coated nanoshells afforded a greater conversion yield than that of related bare palladium nanoshells.
Introduction
The controlled fabrication of nanoscale metallic particles offers the opportunity to develop novel catalysts.1–5 Palladium nanoparticles have received particular attention due to their useful catalytic activities.1,3,6–9 A number of palladium complexes have been developed as homogeneous catalysts for various reactions and have shown good activities and selectivities when compared to heterogeneous analogs.2,10–12 However, for large-scale applications in the solution phase, heterogeneous catalysts are much more attractive because of their ease of separation and recyclability.2,11 Consequently, the crafting of palladium atoms into unique nanoscale metallic particles has been widely studied for the purpose of generating heterogeneous catalysts with unique capabilities.11–13 Due to their large surface-to-volume ratio, nanoscale palladium particles catalyze a diverse scope of reactions, including the hydrogenation of olefins and dienes, the hydration of acrylonitrile to acrylamide, the photogeneration of hydrogen from water, the reduction of carbon dioxide, and a variety of coupling reactions.2,4,8,12,14,15 Interestingly, the reactivity and selectivity of palladium nanoparticles as catalysts depend largely on their size, shape, surrounding medium, and dispersion ability in organic solvents, which facilitates their manipulation and incorporation into various inert supports.16–19Palladium nanoparticles with sizes up to ∼10 nm are routinely prepared by the following methods: sonochemical, gas evaporation, liquid deposition, refluxing alcohol reduction, chemical reduction, and electrochemical.6,20–23 Although these small nanoparticles exhibit excellent catalytic properties, they suffer from a problematic separation from the reaction mixture and are thus not readily amenable to recycling.3,18,24–26 In contrast, a limited number of chemical reduction methods afford large palladium nanoparticles with diameters as large as 50 nm. Typically, these methods involve the reduction of palladium metal ions in the presence of heterogeneous supports or stabilizers such as organic molecules, polymers, and other surfactants.4,7,18,22,27,28
An alternative strategy employs core–shell structures, where the size of palladium nanoparticles can be tuned to greater than 100 nm in diameter in an aqueous solution without using any stabilizers.3,29 Given the stability and dispersity, nanoshells can potentially possess excellent catalytic activity in aqueous or polar organic solutions (e.g., ethanol).3,9 Additionally, the employment of silica and alumina supports (e.g., cores) for palladium-based catalysts has shown enhanced catalytic properties (due to their excellent chemical/thermal stability, good accessibility, and porosity) and the easy recovery of catalysts from a reaction mixture by simple filtration, centrifugation, and decantation.24,25,30–33 This straightforward workup process and simple handling of catalysts is extremely practical for industrial-scale catalysis. For versatile catalytic applications, these palladium nanoparticles/shells on the supports can be further protected with organic molecules to enhance their dispersity in various solvents.8,33–36 Specifically, we propose that coating palladium nanoshells with alkanethiols will enhance their stability and dispersity in organic solvents that can allow for their practical use in numerous catalytic reactions.
Previous research in our laboratory demonstrated that the seeded-growth method can be used to generate palladium shells on silica cores having a variety of diameters.29 Silica nanoparticles are ideal templates for this methodology because of their small polydispersity (less than 1%) and their ease of surface functionalization.37 While our studies did not emphasize the colloidal stability of the palladium nanoshells, we noted that the particles failed to undergo rapid aggregation in an aqueous solution for several weeks. In contrast, it is known that palladium nanoparticles deteriorate rapidly in the solid state, particularly when exposed to reactive gases (e.g., hydrogen).28,38,39
One widely used strategy to stabilize metal nanoparticles is to coat their surface with a self-assembled monolayer (SAM).8,36,39–41 In particular, SAMs derived from alkanethiols are densely packed and defect-free. Consequently, they have the capacity to isolate the particles from the surrounding environment so they have been widely used as protectants for metallic nanoparticles.19,27,34,35,41–44 In the present study, we sought to examine the stabilization of palladium nanoshells by the adsorption of alkanethiols on their surfaces as well as their catalytic efficiency in the Suzuki–Miyaura cross coupling reaction in organic solvents. While it is widely known that good quality SAMs form on flat (2D) palladium substrates,34,40,45,46 there are, to the best of our knowledge, relatively few studies of SAMs on palladium nanoparticles,19,27,28,35,41,47,48 and the particles are quite small (< 10 nm in diameter) compared to those reported here. Notably, these species and related SAM-coated platinum nanoparticles have been shown to be catalytically active despite the presence of the alkanethiol coating, which can offer enhancements in both catalyst stability and catalytic selectivity.19,27,35,41,47–49 In this study, we prepared SAMs of alkanethiols on large palladium nanoshells to investigate their formation and colloidal stability in solution as well as their catalytic activity in various organic solvents.
Experimental section
Materials
All reagents were purchased and used as received from the indicated suppliers: ammonium hydroxide (30% NH3), nitric acid, hydrochloric acid (EM Science), tetraethylorthosilicate, terakis(hydroxymethyl)phosphonium chloride (THPC), 1-dodecanethiol (C12SH), 1-hexadecanethiol (C16SH), 3-aminopropyltrimethoxysilane (APTMS), iodobenzene (all from Aldrich), hydrogen tetrachloroaurate(III) trihydrate (Strem), ethanol (McKormick Distilling Co.), carbon tetrachloride (Mallinckrodt), palladium(II) chloride, L-ascorbic acid, and phenylboronic acid (all from Acros). Deionized water was purified to a resistance of 18 MΩ·cm (Academic Milli-Q Water System; Millipore Corporation) and filtered through 0.22 μm filter membrane to remove any impurities. All glassware and equipment used in the experiments were first cleaned in an aqua regia solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, HCl:HNO3), rinsed in Milli-Q water, immersed in a base bath (saturated KOH in isopropyl alcohol), and then rinsed again in Milli-Q water before use.
:1, HCl:HNO3), rinsed in Milli-Q water, immersed in a base bath (saturated KOH in isopropyl alcohol), and then rinsed again in Milli-Q water before use.
Amine-functionalized silica nanoparticles
Preparation of the amine-functionalized silica nanoparticles (∼100 nm in diameter) was achieved by the modification of the well-known Stöber method.37 Specifically, ammonium hydroxide (6 mL, 28–30% NH3) was added to 100 mL of absolute ethanol in a 250 mL two-necked round-bottomed flask. Tetraethylorthosilicate (3.4 mL, TEOS) was quickly added into the mixture after the solution was warmed to 30 °C. The color of the mixture changed from colorless to milky white within 30 min. The mixture was stirred overnight, and excess APTMS (0.125 mL) was then added to the mixture. The mixture was vigorously stirred with a football-shaped magnetic stirring bar for an additional 6–8 h, and boiled at 88 °C for 1 h to ensure the covalent bonding of APTMS on the surface of the silica nanoparticles.50–52 About 4–5 mL of absolute ethanol was added drop-wise during the refluxing step to maintain the same volume of the mixture. The final solution containing amine-functionalized silica particles was centrifuged in a RC-3B Refrigerated Centrifuge (Sorvall Instruments) at 3000 rpm (revolutions per minute) for 1 h and re-dispersed in 100 mL of ethanol twice to remove any side products, unattached APTMS, and small silica nanoparticles.THPC-reduced Au nanoparticle seeds on amine-functionalized silica nanoparticles
A solution of small gold seeds was prepared by the reduction of hydrogen tetrachloroaurate(III) trihydrate with tetrakis(hydroxymethyl)phosphonium chloride (THPC) using the modification of a Duff et al. method.53,54 A sodium hydroxide solution (1.0 mL of 1 M NaOH), THPC solution (2 mL of water containing 24 μL of 80% THPC), and Milli-Q water (200 mL) were mixed in a 250 mL flask and vigorously stirred for at least 15 min. An aliquot of 1 wt% aqueous HAuCl4·3H2O (4 mL) was quickly added to the reaction mixture. The color of the solution changed quickly from colorless to dark reddish yellow in a few seconds. The mixture was stirred for an additional 30 min. This gold seed solution was aged in the refrigerator for at least three days prior to use.The attachment of gold seed nanoparticles on silica nanoparticles was accomplished by using a slight modification of the Westscott et al. method.55 Briefly, the aged THPC-reduced gold seeds (∼50 mL) were mixed with amine-functionalized silica nanoparticles (1 mL) in a 50 mL centrifuge tube. Unlike other metal nanoshell growth, this seeding process did not require the use of a highly concentrated THPC-reduced gold seed solution. The mixture was shaken for a few minutes and left overnight to ensure the attachment of gold seeds on the silica particles by self-assembly. The mixture was then centrifuged twice for 1 h at 3000 rpm to remove unattached THPC-reduced gold seeds, and the composite particles were re-dispersed in Milli-Q water (50 mL), which afforded a light red solution.
Palladium nanoshell growth
The following is a modification of the original Turkevich et al. method, which was recently used to grow palladium metal layers on silica nanoparticles.7,29 Palladium chloride solution (1 mM) was prepared by dissolving PdCl2 (0.01773 g) in a mixture of water (98 mL) and HCl solution (2 mL at 50 mM). The mixture was aged in a refrigerator overnight. An aliquot of this solution (10 mL) was placed in a 25 mL beaker with a stirring bar, and 3 mL of gold-seeded silica nanoparticles was added to produce palladium nanoshells having consistent Pd shell thicknesses. The mixture was stirred for at least 10 min to mix them completely, and then L-ascorbic acid (0.6 mL of 100 mM) was added quickly. The color changed from yellow to dark steel blue. All samples were centrifuged at 3000 rpm for 30 min, re-dispersed in Milli-Q water twice to remove unreacted chemicals, and stored in a refrigerator before use. Separately, palladium nanoparticles (≤ 10 nm in diameter) were prepared in a similar manner in the absence of silica nanoparticle cores.Alkanethiols on Pd nanoshells by self-assembly (SAMs on Pd nanoshells)
Alkanethiol SAMs were grown on palladium nanoshells using a modification of our previously reported method for growing alkanethiol SAMs on gold nanoshells.43 The palladium nanoshells in Milli-Q water were further centrifuged and re-dispersed in ethanol at least 4 times. Two separate aliquots (25 mL each) of C12SH and C16SH in ethanol (2 mM, 0.35 g and 0.52 g, respectively) were placed in a clean 50 mL beaker containing palladium nanoshells (10 mL: 3 × 1011 particles/mL) in ethanol. Each beaker was covered with aluminum foil and allowed to stir for at least 12 h, leading to the formation of alkanethiol SAMs on the palladium nanoshells. The final solution was centrifuged and re-dispersed in ethanol twice and in CCl4 three times to remove unattached alkanethiols. While bare palladium nanoshells and poorly-coated palladium nanoshells (i.e., incomplete coating of alkanethiols) underwent agglomerations or aggregations, the alkanethiol-coated palladium nanoshells were well-suspended/dispersed in the CCl4 solution.Suzuki–Miyaura coupling using bare Pd nanoparticles/nanoshells and C16SH-Coated Pd nanoparticles/nanoshells3,12,18,26,30,35,56
We examined the Suzuki–Miyaura coupling between phenylboronic acid and iodobenzene using bare and C16SH-modified palladium nanoparticles/nanoshells. Iodobenzene (0.1224 g; 0.600 mmol), phenylboronic acid (0.0744 g; 0.610 mmol), K3PO4 (0.510 g; 2.40 mmol) and ethanol (10 mL) were added to a 50 mL flask. Concentrated bare or C16SH-coated palladium nanoparticles/nanoshells (∼1 mL: 1.5 × 1011 particles/mL in ethanol, ∼0.4 mg of Pd content by ICP-AES analysis) were introduced to the reaction mixture, which was refluxed at 78 °C for 3 h. After cooling the reaction mixture, ∼2 mL diethyl ether was introduced to increase the solubility of the biphenyl product for gas chromatographic (GC) analysis. This Suzuki reaction was also performed in different organic solvents (e.g., toluene and benzene at 80 °C overnight). Since these organic solvents readily dissolve the starting and product molecules, the addition of diethyl ether was not required after the reaction. The resulting mixture was subsequently centrifuged to remove the nanoparticles and K3PO4. The upper solution layer was subjected to analysis by GC. The percent conversion was estimated by examining the peak area ratio between the starting material (iodobenzene was the limiting reagent) and product (biphenyl). All reactions at ≤ 60 °C in organic solvents led to the formation of a trace amount of product (≤ 3%). Although the possible desorption of alkanethiol SAMs from palladium nanoshells was probable under some of our reaction conditions (e.g., the overnight reactions at 80 °C), the GC chromatograms showed no new peaks corresponding to desorbed organosulfur species.Characterization methods
Palladium nanoshells and alkanethiol-coated palladium nanoshells were examined by field emission scanning electron microscopy (FE-SEM), environmental SEM, energy dispersive X-ray (EDX), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FTIR), and ultraviolet-visible (UV-vis) spectroscopy. The catalysis studies were monitored using gas chromatography (GC). Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was also employed to estimate the amount of palladium on the SiO2 cores.A JSM 6330F (JEOL) FE-SEM instrument was employed to examine the morphology and general size distribution of the nanoparticles operating at 15 kV; it was equipped with EDX (Link ISIS software series 300, Oxford Instruments) for elemental analysis. FEI-Quanta 450 SEM instrument (operating at 20 kV) equipped with EDX (X-Max silicon drift detector, 80 mm2, Oxford Instruments, UK) was used for additional imaging and elemental analysis. All samples were deposited onto the carbon-coated TEM copper grid and completely dried at room temperature overnight prior to carbon coating (25 nm in thickness). Similarly, a JEM-2000 FX electron microscope (JEOL) TEM instrument operating at an accelerating voltage of 200 kV was used to examine the core–shell structures. All samples were deposited on 300 mesh Holey carbon-coated copper grids and dried before analysis. An XPS (PHI 5700) system equipped with a monochromatic Al Kα X-ray source (hυ = 1486.7 eV) incident at 90° relative to the axis of hemispherical energy pass analyzer was used to provide an elemental description of the particles. The spectrometer used 23.5 eV of pass energy, 45° of photoelectron takeoff angle from the source, and an analyzer spot diameter of 800 μm. The samples were initially suspended in CCl4, spotted on the surface of the silicon wafer, completely dried, and then extensively cleaned with a high purity N2 gas before analysis. The binding energy was adjusted to 2850 eV (C 1 s) for all the spectra. A Nicolet MAGNA-IR 860 FTIR spectrometer was used to characterize the alkanethiol molecules adsorbed on the palladium nanoshells. The samples were prepared in the same manner as that used in the XPS analyses and were scanned 512 times with background correction at a spectral resolution of 4 cm−1. A Cary 50 Scan UV-visible spectrometer was used to measure the UV-vis spectra ranging from 200 to 1100 nm. All samples were centrifuged at least 3 times to remove unbound alkanethiols, and then re-dispersed in ethanol and transferred into a quartz UV cell.
Gas chromatograms were recorded using a Hewlett Packard 5890 series II gas chromatograph (single injector and single flame ionization detector) equipped with a fused silica capillary column (30 m) and heated gradually to 250 °C (from 150 °C, rate 25 °C min−1) with an inlet pressure of 20 psi. The percent conversion was estimated by measuring the area ratios between the iodobenzene and biphenyl peaks.
To determine quantitatively the amount of palladium metal on the SiO2 nanoparticle cores, an IRIS ICP-AES (Thermo Jarrell Ash Corp.) was employed. An aliquot (1.0 mL) of palladium nanoshells was treated with 1.0 mL of strong acid (1:1 volume ratio HCl and HNO3) in a glass centrifuge tube, and diluted with 9 mL of pure water. The sample solution was centrifuged at 3000 rpm for 30 min to remove the SiO2 cores. The top solution was subjected to ICP-AES analysis. A 100 ppm stock solution of palladium was prepared by adding PdCl2 (17 mg) to 10 mL of the strong acid solution and 90 mL of Nanopure water in a 100 mL volumetric flask. Additional standard solutions were made by diluting aliquots of the stock solution to concentrations of 50 ppm, 25 ppm, 10 ppm, and 1 ppm. A 10% v/v solution of the strong acid in water was used as a blank. The average of three emission intensities at 324.270 nm, 340.458 nm, and 363.470 nm of the samples to the standard curve allowed for the calculation of the content of palladium (∼40 ppm). All values were then collected based on the average of the three measurements.
In separate studies, we used ICP-AES to test for the presence of trace amounts of palladium that could potentially leach from the nanoparticles during the coupling reaction. In these tests, bare palladium nanoshells and C16SH-coated palladium nanoshells were employed in the Suzuki reaction over 12 h in benzene. After reaction, the organic solution was collected and dried, and the residue was re-suspended in water. Analysis by ICP-AES found no palladium in the tested solutions (≪ 1 ppm).
Results and discussion
The seeded-growth method allows for the formation of large and uniform palladium nanoshells consisting of a silica nanoparticle core and a layer of palladium shell.3,29,43 These core–shell nanoparticles are initially stable in aqueous solution, but undergo agglomeration/aggregation in the absence of surfactants. When these nanoshells are exposed to air in the absence of solvents, they readily absorb gaseous molecules (e.g., hydrogen), leading to the possible destruction of the core–shell structure.38,39 However, coating the surface of these metal nanoparticles with self-assembled monolayers (SAMs) increases the colloidal dispersity/stability and inhibits the corrosion of the nanoshell interface.27,57We prepared palladium nanoshells coated with C12SH and C16SH SAMs and analyzed the surface morphologies and size distributions using FE-SEM, DLS, and TEM. Fig. 1a–c show typical FE-SEM images of THPC gold-attached silica nanoparticles, palladium nanoshells on silica cores, and alkanethiol-protected palladium nanoshells. The SEM images and DLS measurements indicate that the size of silica nanoparticle cores is ∼100 nm, while the diameter of the palladium nanoshells is ∼140 nm (both the core and the nanoshells show uniform shapes and narrow size distributions). No significant morphology change was observed before or after the modification of the palladium nanoshells with alkanethiol SAMs. A few small nanoparticles were sometimes visible in the images, which can be attributed to residual palladium nanoparticles generated as side products.
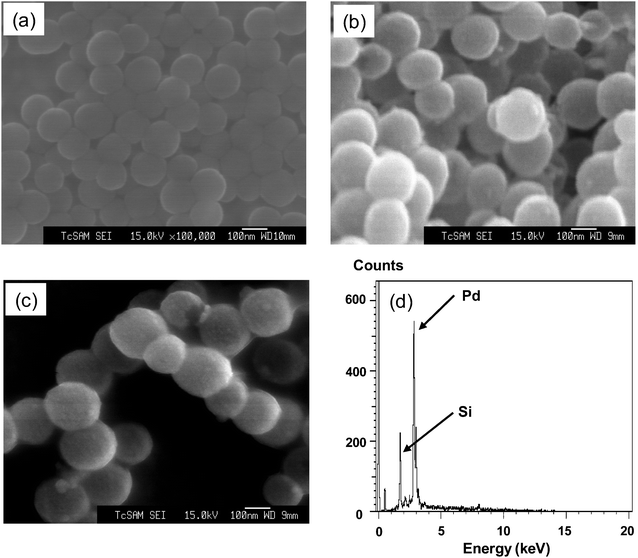 | ||
| Fig. 1 FE-SEM images of the (a) THPC-reduced gold seeds on silica nanoparticles ∼100 nm in diameter, (b) ∼20 nm thick palladium shell-coated silica nanoparticles ∼140 nm in diameter, (c) C16SH-coated palladium nanoshells (∼140 nm in diameter) and (d) EDX spectrum of C16SH-coated palladium nanoshells. | ||
EDX measurements were performed to examine the elemental compositions of the nanoshells. We chose random areas to perform EDX for our samples and observed similar spectra each time. The representative spectrum in Fig. 1d shows an intense peak for palladium shell (Lα, Lβ) at 2.85 keV (∼65 atomic%) and a smaller Si core peak (Kα, Kβ) at 1.75 keV (∼34 atomic%), which suggests the encapsulation of the silica cores by the palladium shells. The small Au peak (Mα) at 2.12 keV probably arises from residual THPC-reduced gold seeds (∼1 atomic%). We observed no obvious peak characteristic of carbon in alkanethiol SAMs, which is probably due to insufficient electrons from the organic molecules. Overall, the EDX data confirm that the nanoshells consist predominantly of Pd and Si.
Fig. 2a shows a TEM image of the surface-tethered silica nanoparticles with THPC-reduced gold seeds. The small dots on the surface of the silica nanoparticles indicate the presence of the attached THPC-reduced gold seeds, which are estimated to be ∼3 nm in diameter. Fig. 2b–d show the growth of the palladium nanoshells on the silica core nanoparticles by notable contrasts in the images coming from the two distinctive electron densities of the core and shell materials. This distinctive contrast allows for the estimation of the dimensions of the palladium shell layer (∼20 nm thick) surrounding the silica nanoparticle core (∼100 nm diameter), which is consistent with the data from the FE-SEM and DLS (not shown). Importantly, the TEM images indicate no clear differences between the bare palladium nanoshells (Fig. 2b) and the C12SH-coated palladium nanoshells (Fig. 2c,d). As a control experiment, we attempted to grow palladium nanoshells directly on amine-functionalized silica nanoparticles in the absence of the THPC-reduced gold seeds on the surface. The lack of gold seeds led to only a slight increase in the diameter of the seeds during the shell growth process (compare Fig. 2a and 2e), indicating incomplete and partially aggregated nanoshell formation. This comparison demonstrates that the gold seeds on the surface of the amine-functionalized silica core play an important role as nucleation sites for the complete and continuous growth of the palladium shell. In a separate study, we were unable to grow palladium nanoshells from palladium seed-attached silica nanoparticle cores, and are still investigating the inefficiency of this particular seed-growth strategy.
 | ||
| Fig. 2 TEM images of the (a) THPC-reduced gold seeds on silica nanoparticles, (b) ∼20 nm thick palladium shell-coated silica nanoparticles, (c) C12SH-coated ∼20 nm palladium nanoshells, (d) C16SH-coated ∼20 nm palladium nanoshells, and (e) (b) palladium shell-coated silica nanoparticles in the absence of THPC-reduced gold seeds. Note the different scale bar for image (d). | ||
Since the SEM and TEM images provided no evidence for the presence of SAMs on the palladium nanoshells, we employed FT-IR and XPS spectroscopy to confirm the attachment of the alkanethiol SAMs on the surface of the nanoshells. The XPS analysis of organic molecules on nanoparticles can often provide useful information regarding the atomic composition of the organic layers and their underlying substrate particles (Fig. 3).34,43,45,58 In particular, XPS can be used to provide information regarding the nature of the sulfur–palladium interaction, and thereby evaluate the covalent attachment of alkanethiols to the surface of the nanoparticles.34,40,45Fig. 3a shows the XPS survey spectrum of C16SH-coated palladium nanoshells with characteristic peaks of Pd3d, C1s, S2p, and Si2p. The presence of a small Au3d peak arising from the THPC gold seeds in the XPS spectrum is consistent with the results of the EDX measurements. From the detailed XPS spectra (Fig. 3b), we observed the binding energy of the S2p3/2 peak at 163 eV. This broad S2p3/2 band consists of two distinctive curve-fitted peaks that are separated by 1.18 eV with a 2:1 area ratio. Since free thiols can be identified by the positioning of the S2p3/2 peak near or above 164 eV, the appearance of the peak at 163 eV confirms the complete binding of the alkanethiols on the palladium nanoshells.34,40,58 Although long alkanethiol SAMs on metal substrates often exhibit slightly higher binding energy due to the insulating effect,43,59 this distinctive phenomenon was not observed for alkanethiol SAMs on the palladium nanoshells. Fig. 3c shows the palladium core-level binding energies at 3d5/2 = 335.3 and 3d3/2 = 340.6 eV for the C12SH-coated palladium nanoshells, and at 3d5/2 = 335.2 and 3d3/2 = 340.5 eV for the C16SH-coated palladium nanoshells. The peak corresponding to palladium appears as a doublet with a splitting of 5.3 eV and an area ratio of 3d5/2:3d3/2 = 4:3, which are in good agreement with the literature.60 These measurements directly confirm the presence of the palladium layers; further, the relatively weak intensity of the Si2p peaks is consistent with the targeted shell/core structure. As a whole, the XPS results are consistent with data obtained with flat palladium surfaces and metal nanoparticles coated with alkanethiols.27,34,40,43
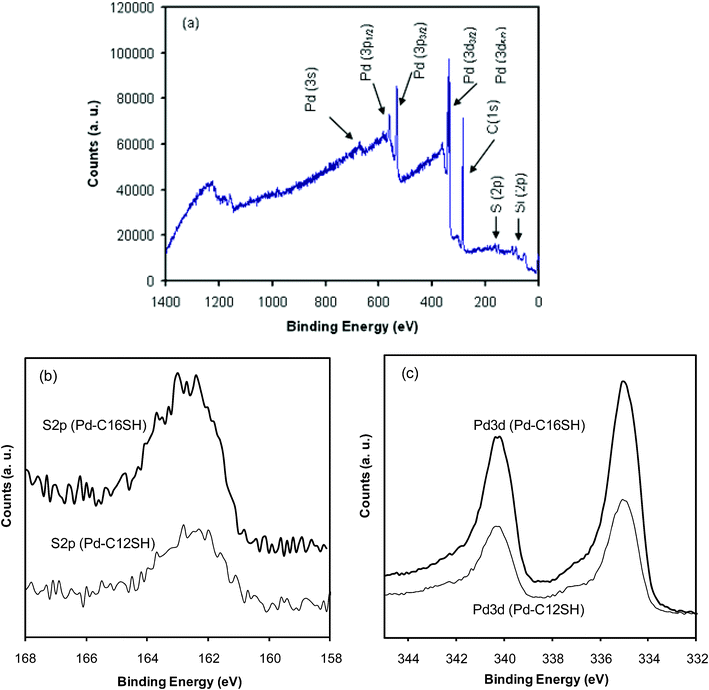 | ||
| Fig. 3 XPS spectra of the (a) survey scan of C16SH-coated palladium nanoshells, (b) S 2p region for C12SH- and C16SH-coated palladium nanoshells, and (c) Pd 3d region for C12SH- and C16SH-coated palladium nanoshells. | ||
FT-IR measurements were performed to evaluate the conformational order of the alkyl chains and thus the packing density of the alkanethiol SAMs (crystallinity of the SAMs) on the palladium nanoshells by examining the carbon–hydrogen stretching region (Fig. 4). FT-IR spectra were obtained in the solid state after the evaporation of the solvent. The band position and width of the C–H symmetric and antisymmetric stretches provide information on the conformational order of the alkyl chains. It is well-known that liquid-state hydrocarbons exhibit a methylene antisymmetric band (νasCH2) at ∼2928 cm−1 and a methylene symmetric band (νsCH2) at ∼2856 cm−1,while crystalline hydrocarbons exhibits them at ∼2918 cm−1 and ∼2850 cm−1, respectively.34,40,50,61 The alkanethiol-coated palladium nanoshells show methylene antisymmetric stretch bands (νasCH2) at 2919–2920 cm−1, indicating the alkyl chains are conformationally ordered (Fig. 4 and Table 1). The crystalline-like conformational order is also supported by the methylene symmetric stretch bands (νsCH2), which appear at 2849–2850 cm−1(Fig. 4 and Table 1). C12SH- and C16SH-coated palladium nanoshells showed no significant differences in conformational order despite the difference in the chain length. The data presented here are again consistent with the data obtained from alkanethiol-coated flat palladium surfaces and other alkanethiol-coated metal nanoparticles.34,40,45,62 In contrast, there is no observation of C–H stretching bands from bare palladium nanoshells (dotted line), confirming that the observed C–H stretches arise from the formation of the alkanethiol monolayer on palladium nanoshells. The formation of highly crystalline organic SAMs in the solid state is also supported by the presence of the wagging progression and twisting-rocking band located at 1375 cm−1 and 1180 cm−1 , as well as the presence of a scissoring band of the methylene groups at 1470 cm−1 for both samples (data not shown).63 Overall, the FT-IR data confirm the formation of well-ordered and densely-packed alkanethiol SAMs on palladium nanoshells.
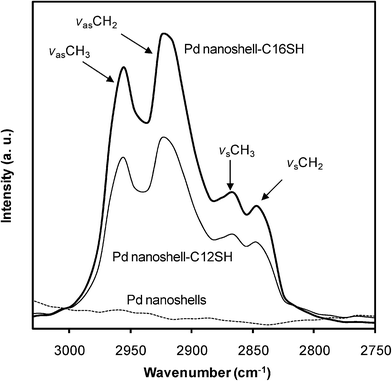 | ||
| Fig. 4 FT-IR spectra of the C–H stretching region of bare palladium nanoshells and alkanethiol-coated palladium nanoshells. | ||
| SAM | Medium | ν s(CH2) | ν as(CH2) | ν s(CH3) | ν as(CH3) |
|---|---|---|---|---|---|
| C12SH | Si wafer | 2849.9 | 2919.2 | 2868.8 | 2954.7 |
| C16SH | Si wafer | 2850.7 | 2920.3 | 2868.9 | 2955.3 |
Alkanethiols, one of the most common chemisorptive surfactants, can effectively stabilize small metal nanoparticles (less than 10 nm) in organic solvents, but normally fail to provide long-term stability to nanoparticles over 10 nm due to the strong van der Waals interactions between large nanoparticles.64 Under our conditions, however, C12SHand C16SH-coated palladium nanoshells over 100 nm in diameter are well-suspended in various organic solvents including hexane, pentane, THF, and CCl4 (Fig. 5). When compared to the bare palladium nanoshells, the solution-phase colloidal stability of the alkanethiol-coated nanoshells supports the formation of organic monolayers on the surface of the nanoshells, which is also confirmed by the XPS and FT-IR data. The alkyl chains of C12SH and C16SH on the palladium nanoshells extend outward, exposing hydrophobic chains at the surface and leading to enhanced dispersion in the hydrocarbon solvents. To evaluate the dispersity, a solution of alkanethiol-coated palladium nanoshells in ethanol was prepared and transferred to many different solvents. The images in Fig. 5 demonstrate that the alkanethiol-coated palladium nanoshells were well-dispersed in all tested solvents except water, while the bare palladium nanoshells displayed notable aggregation, agglomeration, and/or precipitation within a few minutes in CCl4, hexane, pentane, octane, and cyclohexane, as well as exhibiting partial aggregation/agglomeration in other organic solvents such as ether, THF, benzene, chloroform, and dichloromethane.
 | ||
| Fig. 5 Digital images of bare palladium nanoshells and alkanethiol-coated palladium nanoshells in various solvents. | ||
When concentrated bare palladium nonoshells were placed on a solid substrate (e.g., silicon wafer or glass slide for XPS or FT-IR samples) after the evaporation of the mother liquid using a rotary evaporator, we observed a significant increase in the sample size (creamy/foamy) within 2–3 h, probably caused by the absorption of gaseous molecules from the ambient environment.38,39 This volume change of the nanoshells was clearly observable by the naked eye. Under the same circumstances, however, the alkanethiol-coated palladium nanoshells exhibited no similar notable volume changes, even after a week. As such, this simple observation suggests that the formation of alkanethiol SAMs on the surface of palladium nanoshells enhances the stability and/or reduces the process of corrosion in the solid state.
We also investigated the absorption properties of the palladium nanoshells by UV-vis spectroscopy. Fig. 6 shows the UV-vis spectra of the bare and the alkanethiol-coated palladium nanoshells. The samples for these measurements were dispersed in ethanol due to the limited solubility of the bare palladium nanoshells in other organic solvents. The samples were prepared at the same concentration to provide a direct comparison. The bare palladium nanoshells show a weak and broad peak at visible wavelengths and exhibit no distinctive peaks at 235 nm arising from palladium ions, which is consistent with our previous study.29 The entire absorption band of the alkanethiol-coated palladium nanoshells is similar to that of the bare palladium nanoshells, but the absorption maximum appears slightly blue-shifted rather than slightly red-shifted as commonly observed for the adsorption of alkanethiol monolayers on the surface of noble metal nanoparticles.65,66 In our case, it might be that the adsorption of the thiols leads to a smoothening of the typically rough surface generated by ascorbic acid reduction,63,64 which can plausibly give rise to the observed small blue shift. We note, however, that we were unable to detect any changes in size or surface morphology of the nanoparticles upon monolayer formation.
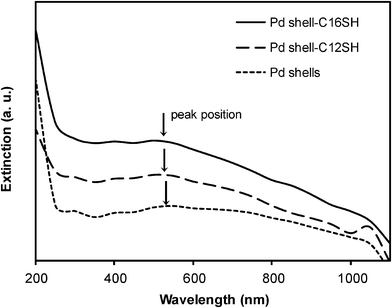 | ||
| Fig. 6 UV-vis spectra of bare palladium nanoshells and alkanethiol-coated palladium nanoshells in ethanol. | ||
Finally, we examined the Suzuki–Miyaura cross-coupling reaction between phenylboronic acid and iodobenzene in the presence of bare and C16SH-coated palladium nanoparticles/nanoshells to examine their catalytic properties in organic media. This coupling reaction was initially performed in ethanol for 3 h using bare palladium nanoparticles (∼60% conversion), bare palladium nanoshells (∼75% conversion), C16SH-coated palladium nanoparticles (∼25% conversion), and C16SH-coated palladium nanoshells (∼50% conversion). When comparing the catalytic activity of the nanoshell and nanoparticle systems, the palladium nanoshells generally afforded slightly higher coupling yields—a result that can perhaps be attributed to a shape-induced enhancement in activity.24,25,32 When comparing the catalytic activity of the bare and the SAM-modified palladium nanoparticles/nanoshells in ethanol, the SAM-modified palladium systems usually gave lower yields, probably due to partial blocking of the catalytic sites by the covalently-bound alkanethiols.35 In contrast, some studies have reported the enhanced catalytic activity of SAM-modified metallic nanoparticle systems by carefully adjusting the structure of the metal cores, the functional group/length of SAM modifiers, and the density of packing,35,47–49 although solubility effects undoubtedly play an important role as well.47–49
Given that the nanoshell system in ethanol afforded slightly higher catalytic activity than the nanoparticle system, we employed bare palladium nanoshells and C16SH-coated palladium nanoshells in the Suzuki reaction in various organic solvents, including benzene, toluene, diethyl ether, and dioxane. In Fig. 7, the GC data obtained in benzene show that the yield of biphenyl using C16SH-coated palladium nanoshells (∼10% conversion in 6 h; ∼20% conversion in 12 h) was slightly higher than that obtained using bare palladium nanoshells (≤ 3% conversion in 6 h; ≤ 7% conversion overnight reaction). The slightly enhanced conversion yield can plausibly be attributed by the higher dispersity/stability of C16SH-modified palladium nanoshells under our reaction conditions. While the reactivity in toluene was comparable to that obtained in benzene, we observed only limited coupling when using diethyl ether and dioxane as the solvents. Notably, the bare nanoshells underwent severe agglomeration/aggregation with excess hydrophilic base (K2PO4) in all of the solvents within 40 min; in contrast, the SAM-modified nanoshells maintained a high dispersity even after 6 h of reaction time at 80 °C (Fig. 7, at the top). Further, increased reaction times in benzene/toluene led to increased degrees of conversion; however, visible agglomeration/aggregation of the SAM-modified nanoshells (i.e., possible desorption of SAMs from nanoshells because of the reaction temperature) was often observed after 8 h. After 12 h of reaction in benzene, the SAM-modified nanoshells and residual base were recovered and used under the same reaction conditions with fresh benzene and reactants. A slight loss in catalytic activity (∼12% conversion after 12 h) was observed. Although the leaching of palladium into solution is possible under our reaction conditions, the ICP-AES studies described in the Experimental Section found no palladium (≪ 1 ppm) in solutions from catalytic runs after 12 h of reaction in benzene using both bare and C16SH-coated nanoshells. These results, however, do not rule out the possibility that palladium can leach from the surface and rapidly redeposit during the reaction. Since we made no attempts to improve the yield of the final product by optimizing the experimental conditions, this preliminary study confirms that the presence of organic SAMs can enhance the stability/dispersity of nanoparticles in various organic media, leading to an increase in the catalytic activity of the nanoshell systems under selected reaction conditions.
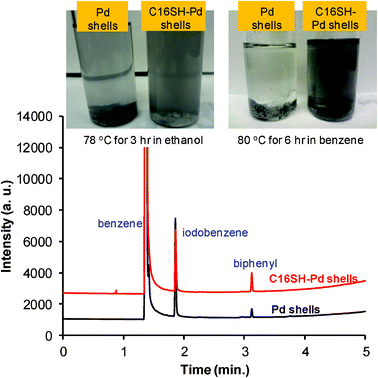 | ||
| Fig. 7 Digital images (top) and gas chromatogram (GC, bottom) of bare palladium nanoshells and C16SH-coated palladium nanoshells after the Suzuki–Miyaura coupling reaction. | ||
Conclusions
We successfully prepared alkanethiol-coated palladium nanoshells by a self-assembly process. The formation of the alkanethiol SAMs on the surface of palladium nanoshells increased the dispersity and colloidal stability of the nanoshells in various organic solvents and in the solid state. Complete analyses using FE-SEM, EDX, TEM, XPS, FT-IR, and UV-vis spectroscopy supported the successful growth of palladium shells on the surface of THPC gold-attached silica nanoparticle cores, as well as the formation of the alkanethiol SAMs on the palladium nanoshells. The alkanethiol monolayers appeared highly ordered and densely-packed on the surface of the palladium nanoshells. A preliminary examination of the use of SAM-coated palladium nanoshells in the Suzuki–Miyaura reaction illustrates the potential for their catalytic applications in various organic solvents. The enhanced dispersity and stability as well as their catalytic activity make it possible to incorporate the palladium nanoshells into diverse media, such as organic solvents, polymers, and porous frameworks.Acknowledgements
We gratefully acknowledge financial support from the National Science Foundation (ECCS-0926027), the Robert A. Welch Foundation (Grant E-1320), the Texas Center for Superconductivity, and the Army Research Office. We thank Dr J. K. Meen for assistance with the FE-SEM measurements and Dr I. Rusakova for assistance with the TEM and EDX measurements. We also acknowledge the College and Chemistry Department of Illinois State University for generous financial support and Dr M. E. Cook for assistance with the additional SEM (NSF DBI-0923448), EDX, and TEM measurements.References
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237 CrossRef CAS.
- H. Hirai and N. Toshima, Tailored metal catalysts, Dordrecht, 1986 Search PubMed.
- S.-W. Kim, M. Kim, W. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS.
- N. Toshima and Y. Wang, Adv. Mater., 1994, 6, 245 CrossRef CAS.
- W. Wang, J. Zhang, F. Chen, D. He and M. Anpo, J. Colloid Interface Sci., 2008, 323, 182 CrossRef CAS.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594 CrossRef CAS.
- J. Turkevich and G. Kim, Science, 1970, 169, 873 CAS.
- Q. Bai, University of Saskatchewan (Saskatoon), 2010.
- F. Wang, C. Li, L.-D. Sun, H. Wu, T. Ming, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2010, 133, 1106 CrossRef.
- R. H. Crabtree, Application to organic synthesis: The organometallic chemistry of the transition metals, 5 ed., Wiley, 2009 Search PubMed.
- D. Astruc, Nanoparticles and catalysis, Wiley, 2007 Search PubMed.
- Y. Li, X. Fan, J. Qi, J. Ji, S. Wang, G. Zhang and F. Zhang, Nano Res., 2010, 3, 429 CrossRef CAS.
- D.-J. Guo and H.-L. Li, J. Colloid Interface Sci., 2005, 286, 274 CrossRef CAS.
- A. J. Bard, Science, 1980, 207, 139 CAS.
- I. Willner, R. Maidan, D. Mandler, H. Dűrr, G. Dőrr and K. Zengerle, J. Am. Chem. Soc., 1987, 109, 6080 CrossRef CAS.
- Y. Mizukoshi, K. Okitsu, Y. Maeda, T. A. Yamamoto, R. Oshima and Y. Nagata, J. Phys. Chem. B, 1997, 101, 7033 CrossRef CAS.
- S. Giorgio, C. Chapon and C. R. Henry, Langmuir, 1997, 13, 2279 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340 CrossRef CAS.
- S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853 CrossRef CAS.
- K. Okitsu, Y. Mizukoshi, H. Bandow, T. A. Yamamoto, Y. Nagara and Y. Maeda, J. Phys. Chem. B, 1997, 101, 5470 CrossRef CAS.
- J. L. Marignier, J. Belloni, M. O. Delcourt and J. P. Chevalier, Nature, 1985, 317, 344 CrossRef CAS.
- G. Gardenas-Trivino, K. J. Klabunde and E. B. Dale, Langmuir, 1987, 3, 986 CrossRef.
- D. Lu and K. Tanaka, J. Phys. Chem. B, 1997, 101, 4030 CrossRef CAS.
- V. Polshettiwara, C. Lenb and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599 CrossRef.
- C. M. Crudden, M. Sateesh and R. Lewis, J. Am. Chem. Soc., 2005, 127, 10045 CrossRef CAS.
- Y. Li, X. M. Hong, D. M. Collard and M. A. El-Sayed, Org. Lett., 2000, 2, 2385 CrossRef CAS.
- S. Chen, K. Huang and J. A. Stearns, Chem. Mater., 2000, 12, 540 CrossRef CAS.
- Y. Li and M. A. El-Sayed, J. Phys. Chem. B, 2001, 105, 8938 CrossRef CAS.
- J.-H. Kim, H.-W. Chung and T. R. Lee, Chem. Mater., 2006, 18, 4115 CrossRef CAS.
- C. Yang, A. K. Manocchi, B. Leeb and H. Yi, J. Mater. Chem., 2011, 21, 187 RSC.
- A. Mieczyńska and A. M. Trzeciak, Molecules, 2010, 15, 2166 CrossRef.
- K. Gude and R. Narayanan, J. Phys. Chem. C, 2010, 114, 6356 CAS.
- N. Kim, M. S. Kwon, C. M. Park and J. Park, Tetrahedron Lett., 2004, 45, 7057 CrossRef CAS.
- J. C. Love, D. B. Wolfe, R. Haasch, M. L. Chabinyc, K. E. Paul, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 2003, 125, 2597 CrossRef CAS.
- F. Lu, J. Ruiz and D. Astruc, Tetrahedron Lett., 2004, 45, 9443 CrossRef CAS.
- R. Tatumi, T. Akita and H. Fujihara, Chem. Commun., 2006, 3349 RSC.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- F. Calvo and A. Carre, Nanotechnology, 2006, 17, 1292 CrossRef CAS.
- M. Moreno, F. J. Ibanez, J. B. Jasinski and F. P. Zamborini, J. Am. Chem. Soc., 2011, 133, 4389 CrossRef CAS.
- G. Corthey, A. A. Rubert, G. A. Benitez, M. H. Fonticelli and R. C. Salvarezza, J. Phys. Chem. C, 2009, 113, 6735 CAS.
- E. Sadeghmoghaddam, C. Lam, D. Choi and Y.-S. Shon, J. Mater. Chem., 2011, 21, 307 RSC.
- L. A. Porter Jr., D. Ji, S. L. Westcott, M. Graupe, R. S. Czernuszewicz, N. J. Halas and T. R. Lee, Langmuir, 1988, 14, 7378 CrossRef.
- T. Pham, J. B. Jackson, N. J. Halas and T. R. Lee, Langmuir, 2002, 18, 4915 CrossRef CAS.
- C. M. Shen, Y. K. Su, H. T. Yang, T. Z. Yang and H. J. Gao, Chem. Phys. Lett., 2003, 373, 39 CrossRef CAS.
- J. C. Love, D. B. Wolfe, M. L. Chabiny, K. E. Paul and G. M. Whitesides, J. Am. Chem. Soc., 2002, 124, 1576 CrossRef CAS.
- T. R. Soreta, J. Strutwolf and C. K. O'Sullivan, Langmuir, 2007, 23, 10823 CrossRef CAS.
- M. Cargnello, N. L. Wieder, P. Canton, T. Montini, G. Giambastiani, A. Benedetti, R. J. Gorte and P. Fornasiero, Chem. Mater., 2011, 23, 3961 CrossRef CAS.
- L. Strimbu, J. Liu and A. Kaifer, Langmuir, 2003, 19, 483 CrossRef CAS.
- S. E. Eklund and D. E. Cliffel, Langmuir, 2004, 20, 6012 CrossRef CAS.
- L. M. Liz-Marzan and P. Mulvaney, J. Am. Chem. Soc., 2003, 107, 7312 CAS.
- A. van Blaaderen and A. J. Vrij, J. Colloid Interface Sci., 1993, 156, 1 CrossRef CAS.
- T. G. Waddell, D. E. Leyden and M. T. DeBello, J. Am. Chem. Soc., 1981, 103, 5303 CrossRef CAS.
- D. G. Duff and A. Baiker, Langmuir, 1993, 9, 2301 CrossRef CAS.
- D. G. Duff and A. Baiker, Langmuir, 1993, 9, 2310 CrossRef CAS.
- S. L. Westscott, S. J. Oldenburg, T. R. Lee and N. J. Halas, Langmuir, 1998, 14, 5396 CrossRef.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 8572 CrossRef CAS.
- F. P. Zamborini, S. M. Gross and R. W. Murray, Langmuir, 2001, 17, 481 CrossRef CAS.
- S. R. Johnson, S. D. Evans, S. W. Mahon and A. Ulman, Langmuir, 1997, 12, 51 CrossRef.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol, K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, Physical Electronics Inc., 1995 Search PubMed.
- R. G. Snyder, H. L. Strauss and C. A. Elliger, J. Phys. Chem., 1982, 86, 5145 CrossRef CAS.
- M. Sastry, Curr. Sci., 2000, 78, 1089 CAS.
- M. J. Hostler, J. J. Stokes and R. W. Murray, Langmuir, 1996, 12, 3604 CrossRef.
- K. Stavens, S. V. Pusztay, S. Zou, R. P. Andres and A. Wei, Langmuir, 1999, 15, 8337 CrossRef CAS.
- A. J. Haes, S. Zou, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. B, 2004, 108, 6961 CrossRef CAS.
- M. D. Malinsky, K. L. Kelly, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2001, 123, 1471 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |