Elucidating active species and mechanism of the direct oxidation of benzene to phenol with hydrogen peroxide catalyzed by vanadium-based catalysts using DFT calculations†
Dianyong
Tang
*ab,
Liangfang
Zhu
c and
Changwei
Hu
*c
aDepartment of Chemistry and Life Science, Leshan Normal College, Leshan, 614000, P. R. China. E-mail: qchem@189.cn; Fax: +86 833 2272106; Tel: +86 833 2272106
bCentre for Molecular Design, Leshan Normal College, Leshan, 614000, P. R. China
cKey Laboratory of Green Chemistry and Technology, MOE, College of Chemistry, Sichuan University, Chengdu, 610064, P. R. China. Fax: +86 28 85411105; Tel: +86 28 85411105E-mail: gchem@scu.edu.cn; chwehu@mail.sc.cninfo.net
First published on 27th January 2012
Abstract
The mechanism by which benzene is converted to phenol through hydroxylation, catalyzed by vanadium in CH3CN is explored at the B3LYP(IEF-PCM)//B3LYP/6-311G(2d,2p) level. Three candidate catalysts are used to simulate the catalytic cycle. The solvent effectively reduces the free energy barriers of the C–H bond activation step. The binuclear vanadium species is predicted to be the main form of the operative catalyst. The cooperative role of the two vanadium centres and the dynamic charge distribution of the binuclear vanadium species are found to increase the catalytic activity. The conservation of aromaticity for the phenyl ring in the benzene or phenyl ligand is essential for the benzene hydroxylation.
Introduction
The direct hydroxylation of benzene into phenol, which involves the activation of a C–H bond and the formation of a new C–O bond, is a challenging topic in chemistry. Recently, it has attracted a great deal of attention.1–5 Over the past few decades, this process has been studied extensively in both liquid and gas phases. Several catalysts, including the Fenton reagent, metal oxides, Pd membranes, ZSM-5 zeolites, and heteropolymolybdates have been investigated.4,6–17 Of these, vanadium and vanadium-containing catalysts have been shown to exhibit high activity.18–29 In our previous studies, sodium metavanadate was found to effectively catalyze the direct hydroxylation of benzene and toluene with hydrogen peroxide in CH3CN medium; we investigated this process using an on-line 51V NMR technique and proposed a suitable mechanism.29 Simultaneously, the mechanism of direct hydroxylation of benzene to phenol with hydrogen peroxide catalyzed by VO2+ in gas phase was studied by using density functional theory (DFT) at the B3LYP/6-311G** level.30 Here, we report a computational study on the mechanism of direct hydroxylation of benzene to phenol with hydrogen peroxide catalyzed by vanadium in CH3CN medium. This investigation fosters the understanding of the active catalyst and interpretation of experimental observations.Computational details
The vanadium species have the following equilibrium values in solvent.31| VO2+ + H2O2 ↔ VO(O2)+ + H2O |
| VO(O2)+ + H2O2 ↔ VO(O2)2− + 2H+ |
| VO(O2)+ + VO(O2)2− ↔ (O2)2V(μ–O)2V(O2) |
| VO2+ + VO(O2)2− ↔ (O2)2V(μ–O)2VO |
VO(O2)+, (O2)2V(μ–O)2V(O2), and (O2)2V(μ–O)2VO can be solvated to form VO(O2)(CH3CN)4+, (O2)2V(μ–O)2V(O2)(CH3CN), and (O2)2V(μ–O)2VO(CH3CN), respectively, in CH3CN solvent. Calculations were performed on the catalytic cycle of benzene to phenol with hydrogen peroxide in CH3CN medium using VO(O2)(CH3CN)4+ (CA1), (O2)2V(μ–O)2V(O2)(CH3CN) (CA2), and (O2)2V(μ–O)2VO(CH3CN) (CA3) as catalysts.
Geometric optimizations and frequency calculations were performed for all stationary points at the density functional level of theory, using the hybrid B3LYP functional and the 6-311G(2d,2p) basis set.32–41 Vibrational analysis was performed to determine the character of each optimized stationary point (minimum or saddle point) and to obtain zero-point vibrational energy values (ZPVE) and thermal corrections under 298.15 K and 1 atm (the experimental conditions). Wave function stability calculations were performed to confirm that the calculated wave functions corresponded to the ground state.42–44
Our calculations were performed in two steps. After optimizing the geometries of intermediates and transition states at the B3LYP/6-311G(2d,2p) level, the effect of the polarized CH3CN environment on the reaction species was evaluated. Self-consistent reaction field (SCRF) single-point energy calculations on the gas-phase-optimized structures in CH3CN continuum (CH3CN as solvent) were carried out using Tomasi's polarized continuum model (IEF-PCM) with the UAHF topological model on the same level as that used for optimization.45,46 As has been demonstrated in other systems, solvation had little effect on geometry.47–49 The differences in electronic energies between the PCM optimized structures and the single-point PCM calculations using the gas-phase geometries were usually less than 2 kcal mol−1. Solvation free energy is the difference between free energy in solution and in the gas phase. Considering the effects of entropy, the following experiments were based on the free energies (ΔG) of activation and reaction. Unless otherwise specified, the natural charges were obtained by natural population analysis (NPA). Natural charges were calculated using natural population analysis at the B3LYP (IEF-PCM)//B3LYP/6-311G(2d,2p) level.50,51
Nucleus-independent chemical shifts (NICS) were computed with the GIAO method at B3LYP/6-311G(2d,2p) level.52,53 NICS provides a practical aromaticity index that can be calculated at the ring centre (non-weighted mean of the heavy atom coordinates on the ring perimeter).
All calculations reported in the present work were carried out with the Gaussian 03 package.54
Results and discussion
Fig. 1 shows the catalytic cycle of the hydroxylation of benzene to phenol catalyzed by VO(O2)(CH3CN)4+ in gas phase and CH3CN solvent at 298.15 K and 1 atm. The optimized structures and selected parameters are shown in Fig. S1 (ESI†). This process includes two elementary reactions, the activation of C–H bond and hydroxyl transfer. First, the coordination of benzene with the vanadium centre of VO(O2)(CH3CN)4+ produces η1-benzene complex IM1, which has an endothermicity of 17.32 and 17.45 kcal mol−1 in the gas phase and CH3CN solution, respectively. The predicted V–C bond length indicates that the interaction between benzene and the vanadium centre is weak and mainly electrostatic (Fig. S1†). Next, phenyl complex IM2 is generated through transition state TS1/2 after activation of the C–H bond with a free energy barrier of 38.04 kcal mol−1 in CH3CN solution. This step is predicted to be endothermic by about 15.13 kcal mol−1 in CH3CN solvent. It involves breakage of the C–H bond and formation of an O–H bond. Orbital analysis makes it clear that the electron of the V–OO bond orbital moves to the σ* anti-bond orbital of the C–H bond to break the C–H bond and form the new V–Cbenzene bond (Fig. S2, ESI†). Then, phenol complex IM3 is formed through hydroxyl transfer with an exothermicity of 79.63 kcal mol−1 in CH3CN solution. The predicted free energy barrier of the hydroxyl transfer step is 37.22 kcal mol−1 in CH3CN solution, which indicates that it is thermodynamically irreversible at room temperature. The occupied molecular orbitals in Fig. S2 (ESI†) clearly show that TS2/3 involves cleavage of the O–O bond and the formation of a new C–O bond. The bond distances are reasonable for the transition state structure, which is responsible for cleavage of the O–O and Cbenzene–V bonds and formation of a Cbenzene–O bond (Fig. S1†). Vanadium plays an important role in the hydroxyl transfer process. Finally, the phenol product is generated from IM3 with the formation (VO2(CH3CN)4+, IM4) of the precursor of the active catalyst. The calculated nuclear independent chemical shifts [NICS(0)]52 of the phenyl ring in the benzene or phenyl ligand of IM1, IM2, and IM3 (Table S1, ESI†) illuminate that the phenyl ring in benzene or the phenyl ligand of IM1, IM2, and IM3 retains its aromaticity. On the basis of the above, it can be said that the rate-determining step of the entire process is the activation of the C–H bond with a free energy barrier of 38.04 kcal mol−1. The high free energy barrier indicates that the hydroxylation of benzene catalyzed by VO(O2)(CH3CN)4+ is not feasible at room temperature.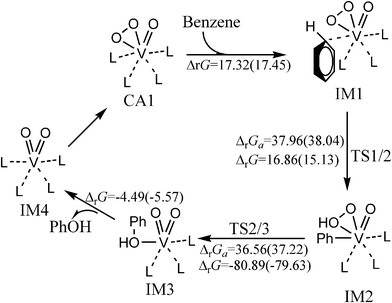 | ||
| Fig. 1 The catalytic cycle and free energy data (units in kcal mol−1) in the gas phase and CH3CN (values in parentheses) with VO(O2)(CH3CN)4+ as catalyst (L = CH3CN) at room temperature. The free energy barriers and reaction free energies are labelled as ΔrGa and ΔrG, respectively. | ||
The catalytic cycle of the hydroxylation of benzene catalyzed by (O2)2V(μ–O)2V(O2)(CH3CN) is shown in Fig. 2. The optimized structures and selected parameters are shown in Fig. S3 (ESI†). The hydroxylation of benzene catalyzed by (O2)2V(μ–O)2V(O2)(CH3CN) has two reaction pathways. The first involves the transfer of hydrogen to O–O on V2 to form an (O2)(OOH)V(μ–O)2V(O2)(phenyl) intermediate. This is called the two-centred pathway. The other involves the transfer of hydrogen to O–O on V1 to produce an (O2)2V(μ–O)2V(OOH)(phenyl) intermediate. This is called the one-centred pathway. The free energy barriers of the activation of the C–H bond and the hydroxyl transfer steps of the two-centred pathway are 28.43 and 24.95 kcal mol−1, respectively, at room temperature.
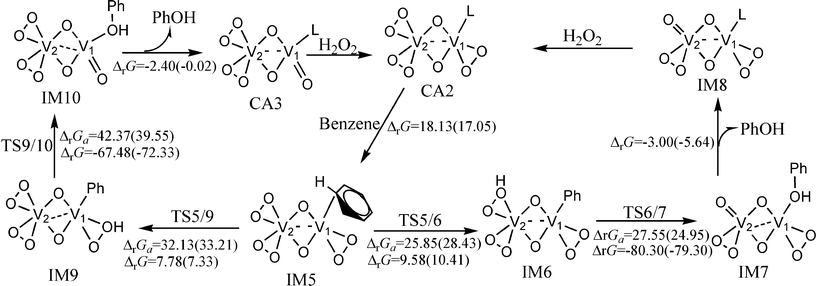 | ||
| Fig. 2 The catalytic cycle and free energy data (units in kcal mol−1) in the gas phase and CH3CN (values in parentheses) with (O2)2V(μ–O)2V(O2)(CH3CN) as catalyst (L = CH3CN) at room temperature. The free energy barriers and reaction free energies are labelled as ΔrGa and ΔrG, respectively. | ||
The hydroxyl transfer step is irreversible at room temperature. The predicted NCIS(0) values of the phenyl rings in the benzene or phenyl of IM5, IM6, and IM7 are −6.52, −9.79, and −9.84, which indicates that the phenyl ring in benzene or the phenyl ligand of IM5, IM6, and IM7 retains its aromaticity. The molecular orbitals of TS5/6 and TS6/7 shown in Fig. 3 indicate that the two-centred pathway involves two vanadium atoms’ cooperative interaction with reaction centres. The transition state (TS6/7) of the hydroxyl transfer step involves the breakdown of Cbenzene–V, O–O, and V2–O bonds, together with the formation of V1–O and Cbenzene–O bonds. The bond distances in Fig. S3 (ESI†) are rational for the said bonding variation. The cooperation of the two vanadium atoms reduces the free energy barrier. For the one-centred pathway, the free energy barriers of the activation of the C–H bond and hydroxyl transfer steps are 33.21 and 39.55 kcal mol−1, respectively. Molecular orbital analysis indicates that these two reactions involve only the V1 atom (Fig. S4, ESI†). The variation of bonding in the hydroxyl transfer step of the one-centred pathway is similar to those of VO(O2)(CH3CN)4+. Comparison of the free energy profiles of the two-centred and one-centred pathways show that the two-centred pathway is more feasible than the one-centred pathway.
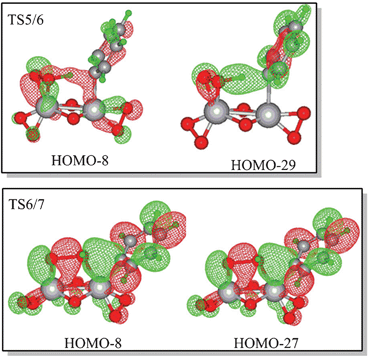 | ||
| Fig. 3 Selected molecular orbitals of the C–H bond activation and hydroxyl transfer transition states of the two-centred pathway with V(O2)2(μ–O)2V(O2)(CH3CN) as the catalyst (cut-off = 0.025). | ||
The catalytic cycle of the hydroxylation of benzene catalyzed by (O2)2V(μ–O)2VO(CH3CN) is similar to that of the two-centred pathway of (O2)2V(μ–O)2V(O2)(CH3CN). It is shown in Fig. 4. Optimized structures and selected parameters are shown in Fig. S6 (ESI†). The free energy barriers of the activation of the C–H bond and hydroxyl transfer steps are 31.43 and 28.10 kcal mol−1, respectively, in CH3CN at room temperature. The molecular orbitals in Fig. S6 (ESI†) show that the orbital interaction in these transition states only involves the V2 centre. The bond distances of reaction centre of TS12/13 are similar to those of TS6/7. The predicted NICS(0) values of the phenyl ring in the benzene or phenyl ligands of IM11, IM12, and IM13 indicate that the aromaticity of the phenyl ring is not destroyed (Table S1, ESI†).
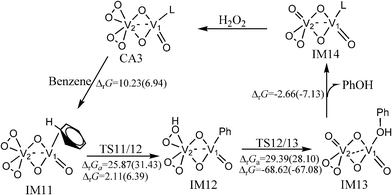 | ||
| Fig. 4 The catalytic cycle and free energy data (units in kcal mol−1) in the gas phase and CH3CN (values in parentheses) with (O2)2V(μ–O)2V(O)(CH3CN) as the catalyst (L = CH3CN) at room temperature. The free energy barriers and reaction free energies are labelled as ΔrGa and ΔrG, respectively. | ||
All of the hydroxylation of benzene to phenol reactions reported herein proceed through the activation of the C–H bond and hydroxyl transfer steps. All of these hydroxylation processes are irreversible. The active order of three catalysts was found to be as follows: (O2)2V(μ–O)2V(O2)(CH3CN) (CA2) > (O2)2V(μ–O)2VO(CH3CN) (CA3) > VO(O2)(CH3CN)4+ (CA1). The binuclear vanadium species (CA2 and CA3) was more stable than the mono-vanadium catalyst (CA1) thermodynamically. CA3 is generated during the hydroxylation of benzene by CA2. For the mono-vanadium catalyst CA1, the rate-determining step was found to be the activation of the C–H bond, with a free energy barrier of 38.04 kcal in CH3CN solution. It was noted that solvent reduces the activation free energy by about 10 kcal mol−1 relative to the hydroxylation of benzene catalyzed by VO(O2)+ in gas phase, as shown in a previous study.30 The interaction between solvent and vanadium was found to be mainly electrostatic. The free energy barrier of the one-centred pathway catalyzed by CA2 was predicted to be 39.55 kcal mol−1. The variation of the charges on V1 and V2 atoms demonstrated that the one-centred pathway involves only the V1 atom, and the V2 atom is a spectator (Table S2, ESI†). However, the activation free energy of the two-centred pathway promoted by CA2 is about 28 kcal mol−1 because two vanadium atoms interact cooperatively with the reaction centres (Fig. 3). For the transition states of the pathway catalyzed by CA3, only the d orbital of the V1 atom participates in orbital interaction. Investigation of the charges on the vanadium atoms and free energy barriers of IM1 → IM2 → IM3 and IM5 → IM9 → IM10 (one-centred pathway of CA2) showed that the increase in the charges of the vanadium atom (V1 in CA2) reduced the free energy barrier of the activation of the C–H bond but increased the free energy barrier of hydroxyl transfer and vice versa (Table S2, ESI†). Simultaneously, charges on the V1 atom are accumulated to decrease free energy barriers of the activation of C–H bond in the activation step of C–H bond, while charges on V1 atom are detract to V2 to make hydroxyl transfer step easy to proceed with the two-centred pathways catalyzed by CA2 and CA3. The dynamic distribution of charges on V1 and V2 atoms in binuclear vanadium species result in the reduction of the free energy barriers (Table S2, ESI†). The bond lengths and bond orders of V1–V2 indicate there is no direct orbital interaction in any of the stationary points on the pathways catalyzed by CA2 and CA3. Throughout the hydroxylation of benzene, the phenyl ring in the benzene or phenyl ligand retains its aromaticity (Table S1, ESI†).
Conclusions
The mechanism for the benzene hydroxylation promoted by vanadium was investigated in a DFT framework at the B3LYP(IEF-PCM)//B3LYP/6-311G(2d,2p) level. Three model catalysts, namely VO(O2) +, (O2)2V(μ–O)2V(O2), and (O2)2V(μ–O)2VO, were investigated separately and compared. Our calculations reveal that the catalytic activity of the hydroxylation of benzene to phenol, for the binuclear peroxovanadium species (O2)2V(μ–O)2V(O2)(CH3CN) and (O2)2V(μ–O)2VO(CH3CN), is higher than that of the monovandium species VO(O2)(CH3CN)4+ because of the cooperative catalysis of the two vanadium atoms of the binuclear peroxovanadium species. The dynamic charge distributions of the two vanadium portions of the binuclear vanadium species increase catalytic activity. (O2)2V(μ–O)2V(O2)(CH3CN) may be the main form of the operative catalyst. The present study shows a clear picture of the mechanism behind the hydroxylation of benzene promoted by vanadium. It supplements our understanding on the active species in the direct hydroxylation of benzene to phenol catalyzed by vanadium-based catalysts.Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (No. 20901053, No. 20502017), the Teaching and Research Award Program for Outstanding Young Teachers in Higher Education Institutions of MOE, P.R.C. (2002), and the Key Project of the Ministry of Education of China (Grant 210189).References
- W. D. Jones, Science, 2000, 287, 1942 CrossRef CAS.
- C. G. Jia, D. G. Piao, J. Z. Oyamada, W. J. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS.
- H. Y. Chen, S. Schlecht, T. C. Semple and J. F.Hartwig, Science, 2000, 287, 1995 CrossRef CAS.
- S. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105 CrossRef CAS.
- R. G. Bergan, Nature, 2007, 446, 391 CrossRef CAS.
- X. Hu, L. Zhu, X. Wang, B. Guo, J. Xu, G. Li and C. Hu, J. Mol. Catal. A: Chem., 2011, 342–343, 41 Search PubMed.
- H. Ehrich, H. Berndt, M. M. Pohl, K. Jahnisch and M. Baerns, Appl. Catal., A, 2002, 230, 271 CrossRef CAS.
- K. Lemke, H. Ehrich, U. Lohse, H. Berndt and K. Jahnisch, Appl. Catal., A, 2003, 243, 41 CrossRef CAS.
- T. Miyahara, H. Kanzaki, R. Hamada, S. Kuroiwa, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2001, 176, 141 CrossRef CAS.
- G. D. Vulpescu, M. Ruitenbeek, L. L. van Lieshout, L. A. Correia, D. Meyer and P. P. A. C. Pex, Catal. Commun., 2004, 5, 347 CrossRef CAS.
- I. Yuranov, D. A. Bulushev, A. Renken and L. Kiwi-Minsker, J. Catal., 2004, 227, 138 CrossRef CAS.
- A. A. Ivanov, V. S. Chernyavsky, M. J. Gross, A. S. Kharitonov, A. K. Uriarte and G. I. Panov, Appl. Catal., A, 2003, 249, 327 CrossRef CAS.
- K. Nomiya, S. Matsuoka, T. Hasegawa and Y. Nemoto, J. Mol. Catal. A: Chem., 2000, 156, 143 Search PubMed.
- K. Nomiya, Y. Nemoto, T. Hasegawa and S. Matsuoka, J. Mol. Catal. A: Chem., 2000, 152, 55 Search PubMed.
- J. Zhang, Y. Tang, G. Li and C. Hu, Appl. Catal., A, 2005, 278, 251 CrossRef CAS.
- N. A. Alekar, V. Indira, S. B. Halligudi, D. Srinivas, S. Gopinathan and C. Gopinathan, J. Mol. Catal. A: Chem., 2000, 164, 181 Search PubMed.
- K. Nomiya, K. Yagishita, Y. Nemoto and T. A. Kamataki, J. Mol. Catal. A: Chem., 1997, 126, 43 CrossRef CAS.
- J. Chen, S. Gao and J. Xua, Catal. Commun., 2008, 9, 728 Search PubMed.
- X. Hu, L. Zhu, B. Guo, Q. Liu, G. Li and C. Hu, Chem. Res. Chin. Univ., 2011, 27, 503 Search PubMed.
- N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358 Search PubMed.
- H. Ge, Y. Leng, F. Zhang, C. Zhou and J. Wang, Catal. Lett., 2008, 124, 250 Search PubMed.
- B. Guo, L. Zhu, X. Hu, Q. Zhang, D. Tong, G. Li and C. Hu, Catal. Sci. Technol., 2011, 1, 1060 RSC.
- Z. Guo, Y. Gu, S. Zhou and C. Ren, Adv. Mater. Res., 2011, 233–235, 1575 Search PubMed.
- X. Gao, X. Lv and J. Xu, Acta Chim. Sin., 2009, 27, 2155 Search PubMed.
- P. K. Khatri, B. Singh, S. L. Jain, B. Sain and A. K. Sinha, Chem. Commun., 2011, 47, 1610 RSC.
- V. Conte and B. Floris, Dalton Trans., 2011, 40, 1419 RSC.
- J. K. Josepha, S. Singhala, S. L. Jaina, R. Sivakumarana, B. Kumara and B. Sain, Catal. Today, 2009, 141, 211 CrossRef CAS.
- Y. Zhu, Y. Dong, L. Zhao and F. Yuan, J. Mol. Catal. A: Chem., 2010, 315, 205 CrossRef CAS.
- M. Jian, L. Zhu, J. Wang, J. Zhang, G. Li and C. Hu, J. Mol. Catal. A: Chem., 2006, 253, 1 CrossRef CAS.
- J. Y. Wang, Ph.D. Dissertation, Sichuan University, Chengdu, 2006 Search PubMed.
- V. Conte, F. D. Furia and S. Moro, J. Mol. Catal., 1994, 94, 323 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1998, 37, 785 CrossRef.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- P. J. Hay, J. Chem. Phys., 1977, 66, 4377 CrossRef CAS.
- K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062 CrossRef.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- R. Seeger and J. A. Pople, J. Chem. Phys., 1977, 66, 3045 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, J. Chem. Phys., 1996, 104, 9047 CrossRef CAS.
- C. Ogretir and I. G. Csizmadia, Computational Advances in Organic Chemistry, Kluwer Academic Publishers, The Netherlands, 1991 Search PubMed.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210 CrossRef CAS.
- D. Tang, L. Zhu and C. Hu, Organometallics, 2011, 22, 5675 Search PubMed.
- M. H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407 CrossRef CAS.
- S. I. Gorelsky, S. Ghosh and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 278 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS.
- NBO Ver. 3.1, E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, Gaussian, Inc., Wallingford CT 2004.
- NICS(0) values were calculated at the centre of the phenyl ring in the phenyl ligand, found by averaging the coordinates of the six carbon atoms forming the ring.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS.
- Gaussian 03, Revision E.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
Footnote |
| † Electronic Supplementary Information (ESI) available: Fig. S1–6 showing molecular orbitals, optimized structures, and selected parameters, Table S1–3 showing NICS(0), natural charges of the vanadium atom, free energies in gas phase, solvation free energies (ΔGsol), and the Cartesian coordinates of the optimized structures. See DOI: 10.1039/c2ra00899h/ |
| This journal is © The Royal Society of Chemistry 2012 |