Green and easy synthesis of biocompatible graphene for use as an anticoagulant†
Yi
Wang
a,
Pu
Zhang
ac,
Chun
Fang Liu
a,
Lei
Zhan
a,
Yuan
Fang Li
a and
Cheng Zhi
Huang
*ab
aEducation Ministry Key Laboratory on Luminescence and Real-Time Analysis, College of Chemistry and Chemical Engineering, Southwest University, 400715, Chongqing, PR China. E-mail: chengzhi@swu.edu.cn; Fax: +86 23 68866796; Tel: +86 23 68254659
bCollege of Pharmaceutical Sciences, Southwest University, 400715, Chongqing, PR China
cCollege of Physical Science and Technology, Southwest University, 400715, Chongqing, PR China
First published on 26th January 2012
Abstract
An environmental friendly method for the preparation of functionalized reduced graphene oxide (rGO) in aqueous solution is developed with great easiness, using heparin as both a reducing agent and an effective stabilizer. Structural and morphological studies have demonstrated that part of the oxygen functionalities in the graphene oxide can be removed by this method, and heparin can be functionalized on the resulting rGO sheets through hydrophobic and hydrogen bonding interactions. The functionalized rGO shows good stability in aqueous solution, owing to the strong electrostatic and steric repulsions of the heparin which adsorb on the surfaces of rGO sheets. Moreover, the rGO exhibits excellent biocompatibility and anticoagulant activity, making it a promising candidate for widespread use in the biomedical field.
Introduction
Graphene, a two-dimensional (2-D) nanostructure with a single-layer of carbon atoms, has attracted tremendous attention in recent years owing to its unique mechanical, thermal, catalytic, electronic and optical properties.1Graphene and graphene-based materials have been applied in a wide range of fields such as biosensing,2drug delivery,3 catalysis,4 and energy storage.5 Thus, how to produce individual graphene sheets in a bulk quantity becomes a significant challenge.Although there has been many methods for the preparation of graphene, solution-based chemical reduction of graphene oxide (GO) to graphene is considered one of the most efficient methods for the low-cost and large-scale production of graphene sheets in the form of monolayers, which can be conveniently deposited on any substrate by simple processes. However, the chemically reduced graphene oxide (rGO) suffers from very limited solubility or even irreversible agglomeration during its preparation in water and most organic solvents, unless capping reagents are used, owing to the strong π–π stacking tendency between rGO sheets. Hence, polymers or surfactants, such as poly (N-vinyl-2-pyrrolidone)6 and poly (sodium-4-styrene sulfonate),7 have been introduced to improve the dispersibility of rGO sheets and prevent them from aggregating. Nevertheless, capping reagents may affect the properties of rGO sheets and be undesirable for practical applications. Moreover, the most used reducing agents, hydrazine and its derivatives, are highly toxic and explosive.8,9 Therefore, precautions must be taken when large quantities of hydrazine or hydrazine derivatives are used, and rGO sheets prepared by this approach may not be fit for use in the biomedical field.
To overcome these shortcomings, several environmentally friendly approaches towards the production of rGO sheets from GO have been developed.8 Biomolecules such as ascorbic acid,10–12amino acids,12,13glucose,14sodium citrate,15 and bovine serum albumin16 have been used as reducing agents or stabilizers. Although the degree of reduction of rGO prepared by these strategies is usually lower than that of the hydrazine-based method, the excellent biocompatibility of these rGO sheets may enhance their feasibilities for use in biological and biomedical fields. However, owing to the insufficient reducing or capping ability of any used “green” reagents for the preparation of rGO, some reported methods suffer from limitations of time-consuming manipulation (e.g., 12–48 h),9,10 relatively poor solubility (e.g., 0.1 mg mL−1),9,11,12 and poor stability (e.g., keep for several days without aggregation, or the stability did not mention in some previous reports)10,11 of the resulting products. These disadvantages may be not propitious to obtain a large quantity of rGO or to store them for a relatively long time. Moreover, the biocompatibility of most final products, biomolecule-functionalized rGO, has not been evaluated,9–15 and some of them also have not been considered for further use.10–12
Heparin, a straight-chain anionic glycosaminoglycan, is widely used as an anticoagulant.17 In the present approach, heparin is utilized as both a reducing agent and a stabilizer to prepare rGO for the following important benefits. Firstly, heparin is a natural polymer material, and has been proven to have excellent biocompatibility.18 Thus, without any other reagents added, the raw material and its reaction products are all environmentally friendly, which will definitely increase the biocompatibility of the obtained rGO. Secondly, the unique chemical structure of heparin (Fig. 1A) makes it not only an ideal reducing agent, but also an effective capping agent. On the one hand, a large number of hydroxyl groups in heparin endow it with strong reducing power. On the other hand, a strong negative charge density from the many carboxylic and sulfonic groups of heparin can be employed to prevent the resulting rGO from aggregation in aqueous solution through electrostatic repulsion interactions, wherein heparin adsorbs on both sides of the surfaces of rGO through noncovalent interactions (e.g., hydrophobic and hydrogen bonding interactions). Thirdly, the resulting heparin-functionalized rGO (heparin-rGO) may exhibit excellent anticoagulant activity, which profits from the unique anticoagulant property of pristine heparin,17 so that it has a potentially wide range of uses in the biomedical field such as for the treatment of thrombosis.
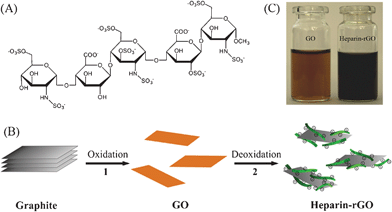 | ||
| Fig. 1 (A) Chemical structure of the repeated units of heparin. (B) Illustration showing the preparation of heparin-rGO in aqueous solution: step 1, oxidative treatment of graphite yields single-layer GO sheets; step 2, chemical reduction of GO by using heparin as both a reducing agent and a stabilizer to produce a stable aqueous suspension of heparin-rGO sheets. (C) Photograph of aqueous dispersions of GO (brown) and heparin-rGO (black). | ||
Therefore, an easy and “green” approach for preparing rGO in aqueous solution is proposed, using heparin as both a reducing agent and a stabilizer (Fig. 1B). The present method is simple, efficient, environmentally friendly, and a large quantity of heparin-rGO with good stability in aqueous solution can be obtained. Furthermore, macroscopic heparin-rGO paper can be easily fabricated through the vacuum filtration method. Most importantly, the as-prepared heparin-rGO sheets show excellent biocompatibility and activity for blood anticoagulation, making it a promising material for biomedical applications.
Experimental section
Materials
Heparin (185 USP units/mg) was purchased from Aladdin (Shanghai, China). Graphite powder was supplied by the Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Cell Counting Kit-8 containing WST-8 reagent was purchased from Beyotime Institute of Biotechnology (Jiangsu, China) for evaluating the cell growth inhibition assays. Reagents for the blood coagulation studies, including the indicators of activated partial thromboplastin time (APTT) and prothrombin time (PT), were ordered from SUNBIO Biotechnology Co., Ltd (Shanghai, China). Doxorubicin hydrochloride was supplied by the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Mili-Q purified water (18.2 MΩ) was used throughout the experiments.Characterization
Ultraviolet-visible (UV-vis) absorption spectra were measured with a 3600 UV-visible-NIR spectrophotometer (Shimadzu, Japan). A FTIR-8400S Fourier transform infrared (FT-IR) spectrophotometer (Shimadzu, Japan) was used for recording IR spectra. Raman spectra were obtained on a InVia Raman spectrometer (Renishaw, England) with 514.5 nm wavelength incident laser light. An S-4800 scanning electron microscope (Hitachi Ltd., Japan) was used to record the scanning electron microscopy (SEM) images. Atomic force microscopy (AFM) images were recorded using a Multimode Nanoscope TM scanning probe microscopy system (Veeco, USA). X-Ray diffraction (XRD) patterns were measured on an XD-3 X-ray diffractometer (Beijng, China) using Cu Kα radiation (λ = 1.5406 Å). Dynamic light scattering (DLS) and ζ-potential measurements were performed using a Zetasizer Nano-ZS90 System (Malvern Inc.). The photographs of solutions were captured with an Olympus E-510 digital camera. The cell livability was measured by Mode 680 Microplate Reader (BIO-RAD, USA). CoaDATA2001 semi-automatic blood coagulation analyzer (LABITEC, Germany) was used to evaluate the anticoagulant activity of graphene.Preparation of heparin-rGO and its paper
GO sheets were synthesized by a modified Hummers method.19,20 The experimental details are shown in the ESI.† Using GO as a precursor, heparin-rGO sheets were prepared using heparin as both a reducing agent and a stabilizer. In a typical procedure, 100 mg heparin was added into 50 mL of homogeneous GO dispersion (0.2–1.0 mg mL−1), followed by ultrasonic irradiation for 1 h. Then, to the resulting dispersion were added 50 μL of ammonia solution (28% w/w) to adjust the pH and promote the reducing reaction. After being vigorously stirred for 5 min, the mixture was stirred in a water bath (95 °C) for 1 h. The resulting stable black dispersion was then centrifuged and washed with water three times. Finally, the obtained heparin-rGO sheets were redispersed in water before further use.Macroscopic heparin-rGO paper was fabricated by the vacuum filtration method.21 In brief, the heparin-rGO dispersion was filtered through a cellulose acetate membrane (47 mm in diameter, 220 nm pore size) by vacuum at room temperature. Followed by air drying, the rGO paper could be easily peeled off from the substrate of the membrane. The thickness of the rGO paper could be controlled by adjusting the concentration and volume of the suspension. The rGO paper was cut by a razor blade into rectangular strips for the cross-sectional view by SEM imaging.
Cell growth inhibition assays of heparin-rGO
Hela cells were used to reveal the biocompatibility of GO, heparin-rGO and hydrazine-reduced rGO (hydrazine-rGO). Firstly, a series of concentrations (1−65 μg mL−1) of GO, heparin-rGO and hydrazine-rGO were incubated with cells in 96-well plates for 24 h (37 °C), respectively. Then, the cells were washed by phosphate buffer three times, and WST-8 reagent was added and incubated with them for 0.5 h. Finally, the data were recorded by the Mode 680 Microplate Reader, and the cell livability was calculated according to the absorbance.Anticoagulant activity investigations of heparin-rGO
Plasma preparation: nine volumes of blood from healthy volunteers were mixed with one volume of sodium citrate (0.109 mol L−1). Then, the plasma with yellow color was obtained immediately by the centrifugation of the mixture (3000 r/min, 15 min). The citrated plasma was used in 2 h in room temperature, or could be kept in −20 °C for 24 h.Measurement of APTT: appropriate amounts of GO, heparin-rGO, and hydrazine-rGO were added with 0.1 mL APTT reagent to cups and warmed for 1 min at 37 °C, respectively. Citrated plasma (0.1 mL) was added to the cups and the mixtures were incubated at 37 °C for 5 min. Then, 0.1 mL of CaCl2 (0.025 mol L−1) was added to recalcify the citrated plasma, and the clotting time was measured. Each determination was performed in triplicate and the average value was obtained.
Measurement of PT: appropriate amounts of GO, heparin-rGO, and hydrazine-rGO were added with 0.2 mL of PT reagent to cups and warmed for 1 min at 37 °C, respectively. Then, citrated plasma (0.1 mL, warmed for 1 min at 37 °C) was added to the mixture, and the clotting time was measured. Each determination was performed in triplicate and the average value was obtained.
Results and discussion
Preparation and stability of heparin-rGO
As illustrated in Fig. 1B, heparin-rGO was synthesized by a two-step approach including an oxidation step and a heparin-based reduction step. In step 1, water-soluble GO sheets with brown color (Fig. 1C) were obtained by the oxidation of graphite according to a modified Hummers method.19,20 Polar functional groups, such as hydroxyl, epoxyl and carboxyl, were introduced into the inert graphene sheets. In step 2, a chemical deoxidization process of GO was carried out to form heparin-rGO by using heparin as both a reducer and a stabilizer. In that way, heparin could be adsorbed on the surfaces of the resulting rGO sheets, and prevent them from aggregation by a strong electrostatic repulsion interaction, which was derived from the high negative charge density of its plentiful sulfonic and carboxylic groups. Finally, a stable black aqueous suspension of heparin-rGO sheets could be obtained (Fig. 1C).UV-vis absorption spectroscopy was first used to confirm the reduction of oxygen-containing groups in GO by heparin. As shown in Fig. 2A, the absorption peak of GO dispersion located at 227 nm with a shoulder peak at about 300 nm, which was consistent with the previous reports.10,14 After the deoxidization process finished, the peak red-shifted to 255 nm and the absorbance increased dramatically in the whole spectral region (> 220 nm). This result suggested that GO was reduced by heparin and the aromatic structure of graphene might be restored. In addition, we also studied the absorption spectrum of hydrazine-rGO in parallel for comparison, which showed its peak at 265 nm. The absorption peak of GO shifts to a longer wavelength after the reduction process, and the shift increases with the increase of the degree of reduction. Therefore, it was demonstrated that the ability of heparin for reducing GO was relatively weaker than that of hydrazine.
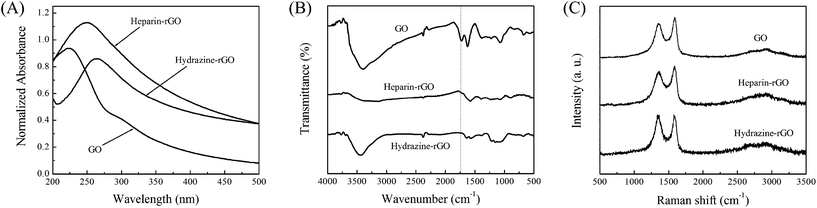 | ||
| Fig. 2 Spectral characterization of GO, heparin-rGO and hydrazine-rGO: (A) UV-vis absorption spectra, (B) FT-IR spectra, and (C) Raman spectra. | ||
Further characterization by FT–IR spectroscopy was performed to evaluate the deoxidization of GO by heparin. As shown in Fig. 2B, GO displayed the characteristic FT-IR peaks corresponding to the oxygen functionalities, such as the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak at 1724 cm−1, the C–O (epoxy) stretching vibration peak at 1227 cm−1, the C–O (alkoxy) stretching peak at 1072 cm−1, and the vibration and deformation peaks of O–H groups at 3399 cm−1 and 1385 cm−1, respectively. After 1 h reaction of the GO with heparin, these peaks decreased dramatically, and even disappeared entirely (e.g., 1724 cm−1). These changes of heparin-rGO compared with GO in FT-IR spectra were obvious, which was identical with that of hydrazine-rGO. Thus, it confirmed that some of the oxygen functionalities in the GO could be removed by heparin.10
O stretching vibration peak at 1724 cm−1, the C–O (epoxy) stretching vibration peak at 1227 cm−1, the C–O (alkoxy) stretching peak at 1072 cm−1, and the vibration and deformation peaks of O–H groups at 3399 cm−1 and 1385 cm−1, respectively. After 1 h reaction of the GO with heparin, these peaks decreased dramatically, and even disappeared entirely (e.g., 1724 cm−1). These changes of heparin-rGO compared with GO in FT-IR spectra were obvious, which was identical with that of hydrazine-rGO. Thus, it confirmed that some of the oxygen functionalities in the GO could be removed by heparin.10
Raman spectroscopy was also utilized to study the structure of heparin-rGO. Pristine graphite displayed a prominent G peak at 1580 cm−1 (ESI,† Fig. S1), corresponding to the first-order scattering of the E2g mode.22 However, the Raman spectra of GO displayed two prominent peaks at 1589 and 1357 cm−1 (Fig. 2C), which correspond to the well-documented G (the E2g mode of sp2carbon atoms) and D (the symmetry A1g mode) bands, respectively.10,22 The D band became prominent indicating the reduction in size of the in-plane sp2 domains, owing to the extensive oxidation of graphite. These bands still existed in the Raman spectra after the reduction of GO by heparin, but the ratio of D/G intensity increased compared with that of GO. This change in Raman spectra was identical with hydrazine-assisted reduction of GO, which confirmed the deoxidation ability of heparin toward GO.
To evaluate the stability of heparin-rGO sheets in aqueous solution, the measurement of ζ-potential was carried out. Fig. 3B showed that an average ζ-potential of GO with –40.7 ± 0.57 mV was found, indicating that large numbers of carboxyl groups with highly negative charge density were formed in the GO sheets.23 This value decreased to –51.3 ± 0.46 mV after the reduction of GO by heparin. The trend of ζ-potential data proved that the heparin-rGO possessed more surface negative charge density than GO, since heparin functionalized on the surfaces of rGO sheets and introduced a large number of negative charges. The electrostatic repulsion interaction of such negative charges was important for the stable dispersion of graphene sheets in aqueous solution.23,24 According to the definition of colloid stability with ζ-potential by the American Society for Testing and Materials,25 our heparin-rGO aqueous dispersion showed “good stability” as its ζ-potential was lower than −40 mV. Thus, there was no sign of aggregation of heparin-rGO sheets after more than three months in aqueous solution.
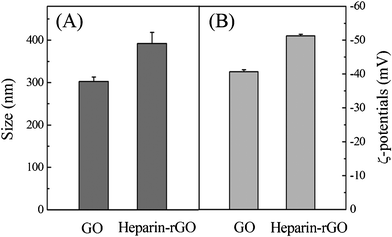 | ||
| Fig. 3 DLS (A) and ζ-potential (B) measurements of GO and heparin-rGO. | ||
Furthermore, DLS measurement was performed to elucidate the state of heparin-rGO sheets in aqueous solution. It was found that the average hydrodynamic diameter (AHD) of GO was 302.5 ± 10.6 nm (Fig. 3A). However, after the reduction of GO with heparin, an AHD of 392.4 ± 26.1 nm was obtained under the same instrumental conditions, which was relatively larger than that of GO. This obvious change of size distribution indicated that heparin not only acted as a reducing agent to prepare rGO, but also was functionalized on the surfaces of the resulting rGO, leading to a decreased Brownian motion rate after the reduction process. Since the particles were assumed to be spherical in the DLS analysis, the data in our case could not be taken to evaluate the actual sizes of the 2-D structures, and could only be used to reflect the trend of changes in the graphene's size.23,26,27
Structure studies of heparin-rGO
The structure of heparin-rGO was further studied by AFM, SEM, and XRD. Samples for AFM measurements were prepared by drop-casting the corresponding dispersions onto freshly cleaved mica substrates. Then, cross-sectional AFM images and the corresponding height measurements of single-layered GO and heparin-rGO were recorded (Fig. 4). The average thickness of the single-layer GO sheet was 1.0 nm, which was consistent with the value reported in the previous literature.14 Nevertheless, the average thickness increased to 1.3 nm after the GO was reduced and functionalized with heparin. This result revealed that the capping reagent, heparin, played an important role in increasing the thickness of the resulting heparin-rGO. In addition, from the three-dimensional (3-D) images (ESI,† Fig. S2), it was found that the surfaces of GO were flat, while the peripheral areas of heparin-rGO were more likely thicker than that of centric areas. This observation suggested that heparin was able to coat the peripheral areas of rGO, which might be attributed to the strong hydrogen bonding interactions between the residual peripheral carboxyl of rGO and heparin.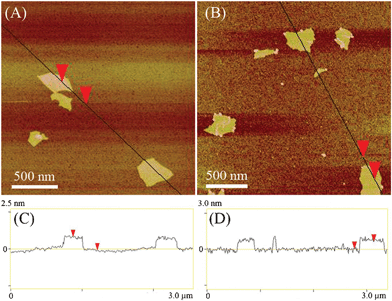 | ||
| Fig. 4 AFM images and the corresponding height measurements of GO (A and C) and heparin-rGO (B and D). | ||
SEM images of GO and heparin-rGO sheets were also recorded (ESI,† Fig. S3). Because the monolayer graphene sheets were extremely thin and almost transparent, it was hard to make a distinction between them and the silicon substrates. However, crumpled silk waves and edges of these carbon sheets made us believe that monolayer GO and rGO sheets were formed, which is consistent with the observation of a previous report.28
To further characterize their crystal structures, XRD patterns of the pristine graphite, exfoliated GO, and heparin-rGO were studied (Fig. 5). Compared with the pristine graphite (2θ = 26.4°), the diffraction peak of exfoliated GO moved to 10.4° (002) with the layer-to-layer distance (d-spacing) of 0.85 nm. This value was larger than the d-spacing of pristine graphite (0.34 nm), as a result of the introduction of numerous oxygenated functional groups on the carbon sheets. After the exfoliated GO was reduced by heparin, the peak at 10.4° disappeared, but a new diffraction peak appeared (2θ = 22.9°, d-spacing 0.39 nm), which was closer to the typical (002) diffraction peak of graphite (2θ = 26.4°, d-spacing 0.34 nm). In addition, the XRD pattern of hydrazine-rGO was also measured in parallel for comparison (ESI,† Fig. S4), which showed the main peak at 25.2° (d-spacing 0.35 nm). This result further confirmed the reduction of GO by heparin.10,14,15
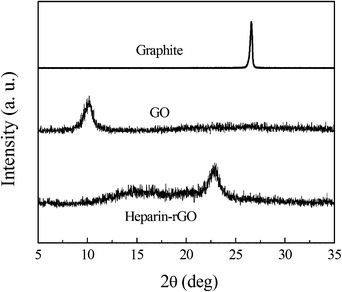 | ||
| Fig. 5 XRD patterns of pristine graphite, GO and heparin-rGO. | ||
Uniform graphene papers also could be easily fabricated by the vacuum filtration of as-prepared heparin-rGO dispersion.21 The thickness of the papers could be adjusted by the volume and concentration of the heparin-rGO dispersion, and free-standing graphene papers could be easily peeled off from the membrane substrate. As shown in Fig. 6A, the obtained graphene paper exhibited good flexible and a shiny lustre. Moreover, the cross-sectional view of the heparin-rGO paper imaged by SEM clearly indicated its well-packed layered structure (Fig. 6B). Such heparin-rGO paper can be easily obtained for mass production, and may be a potential candidate for practical applications.
 | ||
| Fig. 6 Photograph of heparin-rGO paper prepared by vacuum filtration method (A) and SEM image of its transverse section (B). | ||
Mechanism of forming heparin-rGO
The results of spectral and structural investigations indicated that heparin could not only act as an efficient reducing agent, but also play an important role in stabilizing the resulting rGO sheets. Heparin, a highly-sulfated anionic glycosaminoglycan, has the highest negative charge of any known biomolecule.29 On the one hand, a large number of hydroxyl groups in its repeating saccharide units make heparin have strong reducing ability, and the oxygenated functional groups in GO sheets can be easily chemically reduced. On the other hand, compared with the pendent carboxylic and sulfonic groups of heparin, its backbone is relatively hydrophobic. Thus, the hydrophobic interaction between the as-prepared rGO and heparin is effective for their noncovalent conjugation. Moreover, strong hydrogen bonding interactions between the residual carboxyl of rGO and hydroxyl as well as carboxyl groups of heparin or its oxidized products also contribute to the formation of heparin-rGO composite, which was supported by the AFM images in 3-D mode (ESI,† Fig. S2). Such a mechanism is similar to the previous report.14 Once the heparin-rGO forms, the strong electrostatic and steric repulsion interactions can disrupt the π–π stacking interaction between the rGO sheets that prevent them from aggregation. Therefore, the designed heparin-rGO can be prepared easily, and be stable for more than three months in aqueous solution.Biocompatibility investigation of heparin-rGO
In order to evaluate the biocompatibility of as-prepared heparin-rGO, the toxicological effect against Hela cells was investigated. As shown in Fig. 7, the livability of Hela cells which were incubated with GO was always around 100% under the used concentrations (1–65 μg mL−1) after 24 h exposure. This result indicated that GO was quite biocompatible even if relatively high concentrations were used, which confirmed the report from previous literature.30 Delightedly, our prepared heparin-rGO displayed similar cell livability to GO, because the cell livability was always above 95% when the concentrations of heparin-rGO used were in the range 1–65 μg mL−1. It suggested that highly biocompatible rGO could be obtained by the present method. At present, the most general wet-chemical method to prepare rGO is hydrazine-based reduction of GO. Thus, the biocompatibility of hydrazine-rGO was also investigated in parallel as a control. It was found that the cell livability decreased gradually with the increase of the concentration of hydrazine-rGO. When the hydrazine-rGO with a concentration of 65 μg mL−1 was used to incubate with Hela cells, the livability decreased to about 40%, which was distinct with that of GO and heparin-rGO. This result demonstrated that hydrazine-rGO is more toxic than the other two kinds of graphene, owing to the high toxicity of the reducing agent, hydrazine. Previously, the toxicological studies in vitro also demonstrated that the hydrazine-rGO was highly toxic to cells.30 Therefore, it was considered that the surface chemistry was the primary contributor to the difference of toxicity between hydrazine-rGO and heparin-rGO. Obviously, the present method and the corresponding resulting products were environmentally friendly, which was attributed to the impeccable biocompatibility of heparin.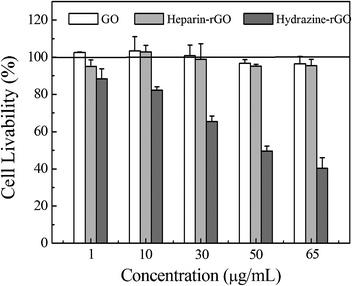 | ||
| Fig. 7 Cell growth inhibition assays of GO, heparin-rGO, and hydrazine-rGO at various concentrations, respectively. | ||
Anticoagulant activity of heparin-rGO
The ultimate goal of preparing multifarious functionalized graphene is to endow it with unique prospects for further use. Here, owing to its excellent biocompatibility, the heparin-rGO is propitious to be applied in the biological and biomedical fields. Thus, the biological activity of heparin-rGO was further investigated. It was found that the as-prepared heparin-rGO exhibited excellent activity as a blood anticoagulant. As shown in Fig. 8A, the citrated plasma (left tube) which was used as a control coagulated in 30 min after adding an appropriate volume of CaCl2 (0.025 mol L−1). However, another citrated plasma that was treated with heparin-rGO beforehand (right tube) could not coagulate in 30 min after adding the same volume of CaCl2 (0.025 mol L−1). Moreover, this sample could not coagulate even after 1 h. The prolongation in recalcification time of citrated plasma treated with heparin-rGO suggested that heparin-rGO could play an important role in the blood coagulation process.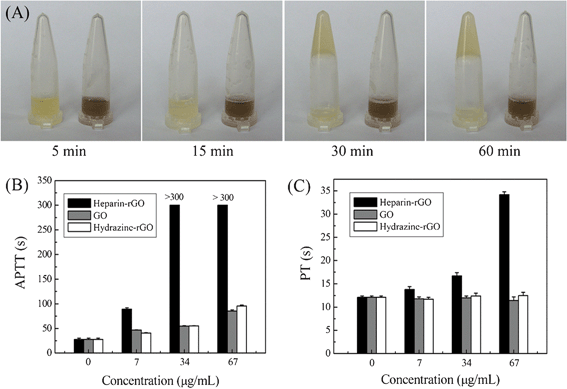 | ||
| Fig. 8 Influence of graphene on the blood coagulation tests: (A) photographs of the plasma without (left)/with (right) heparin-rGO at different times; (B) APTT tests of the plasma added with different types of graphene including heparin-rGO, GO, and hydrazine-rGO, respectively; (C) PT tests of the plasma added with heparin-rGO, GO, and hydrazine-rGO, respectively. | ||
Blood clotting is the result of a complex process initiated by the intrinsic system or the extrinsic system and/or a common pathway.31 In order to further clarify the anticoagulant mode of heparin-rGO, the blood coagulation indicators of APTT and PT were measured. GO and hydrazine-rGO sheets were analyzed in parallel as a control (the details for their synthesis are shown in the ESI†). As shown in Fig. 8B, compared with the APTT value of healthy human plasma (27.7 ± 2.5 s), heparin-rGO prolonged APTT and made it reach 89.4 ± 2.4 s at a concentration of 7 μg mL−1. Moreover, the values of APTT increased remarkably with increasing the concentrations of heparin-rGO, and even exceeded 300 s with the concentrations of 34 μg mL−1 and 67 μg mL−1, respectively. In addition, GO and hydrazine-rGO with the same concentrations also could prolong the APTT, but the effects could not compare with that of heparin-rGO. This result indicated that the heparin, which had been confirmed to coat on the surfaces of the obtained rGO, played an important role in the contribution of the anticoagulant activity of heparin-rGO composite. Similarly, compared with the PT value of healthy human plasma (12.1 ± 0.3 s), heparin-rGO prolonged it to 13.8 ± 0.6 s at 7 μg mL−1, 16.7 ± 0.7 s at 34 μg mL−1, and 34.2 ± 0.6 s at 67 μg mL−1, respectively (Fig. 8C). On the contrary, GO and hydrazine-rGO showed no prolongation effect on PT at the test concentration range.
As the various coagulation assays indicate the interactions with different pathways of the blood coagulation, they can provide basic information about the mode of action of anticoagulants. APTT is used for the evaluation of the intrinsic pathway of blood coagulation, while PT is used for the evaluation of extrinsic pathway in clinical medicine.31 Heparin-rGO can prolong both APTT and PT, indicating that the inhibition of common pathways (intrinsic and extrinsic pathways) of blood coagulation by our prepared heparin-rGO can be convinced. However, the prolongation of APTT but no effect on the PT of GO and hydrazine-rGO suggests that they can only inhibit the intrinsic pathway of coagulation. Moreover, the effect on the APTT of GO and hydrazine-rGO is much weaker than that of our prepared heparin-rGO. Overall, the results suggest that graphene has significant anticoagulant activity, and the inhibitory pathways of blood coagulation are different with different kinds of graphene. The initial results reported here are promising, but more significant work is needed to further understand the anticoagulant mechanism of graphene.
Conclusions
We developed an environmentally friendly and easy method for the preparation of heparin-rGO, wherein heparin not only acts as an effective reducing agent, but also plays an important role as a capping reagent in stabilizing the resulting rGO. Structural and morphological studies demonstrated that the oxygen functionalities in the GO could be removed by heparin without using any other reducing agents, and synchronously, heparin could be functionalized on the resulting rGO sheets through hydrophobic and hydrogen bonding interactions. Highly negative charge density of heparin could prevent the resulting rGO from aggregation in aqueous solution by strong electrostatic repulsion. Therefore, large-scale heparin-rGO could be obtained and stably dispersed in water for more than three months. Macroscopic heparin-rGO paper could be easily fabricated by the vacuum filtration method. Furthermore, the as-prepared heparin-rGO showed excellent biocompatibility to cellsin vitro. Blood anticoagulation investigations revealed that the heparin-rGO could inhibit both intrinsic and extrinsic pathways of coagulation, while GO and hydrazine-rGO could only affect the intrinsic pathway. In addition, the present heparin-rGO could strongly bind to anti-cancer drugs for highly efficient loading (ESI,† Fig. S5), which might combine with its advantages of biocompatibility and anticoagulant activity, making it a promising candidate for widespread use in the biomedical field.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21035005), Postgraduate Science and Technology Innovation Program of Southwest University (No. ky2009004), Fundamental Research Funds for the Central Universities (No. XDJK2010D001), and Large Instruments and Equipment Fund of Southwest University.References
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS.
- H. Cheng, X. Sha, L. Chen, A. C. Cooper, M.-L. Foo, G. C. Lau, W. H. Bailey and G. P. Pez, J. Am. Chem. Soc., 2009, 131, 17732–17733 CrossRef CAS.
- X.-Z. Tang, Z. Cao, H.-B. Zhang, J. Liu and Z.-Z. Yu, Chem. Commun., 2011, 47, 3084–3086 RSC.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- J. I. Paredes, S. Villar-Rodil, M. J. Fernández-Merino, L. Guardia, A. Martínez-Alonso and J. M. D. Tascón, J. Mater. Chem., 2011, 21, 298–306 RSC.
- D. Y. Lee, Z. Khatun, J.-H. Lee, Y.-k. Lee and I. In, Biomacromolecules, 2011, 12, 336–341 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- M. J. Fernandez-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. D. Tascon, J. Phys. Chem. C, 2010, 114, 6426–6432 CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and L. Niu, Langmuir, 2009, 25, 12030–12033 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS.
- Z. Zhang, H. Chen, C. Xing, M. Guo, F. Xu, X. Wang, H. J. Gruber, B. Zhang and J. Tang, Nano Res., 2011, 4, 599–611 CrossRef CAS.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS.
- G. M. Arepally and T. L. Ortel, Annu. Rev. Med., 2010, 61, 77–90 CrossRef CAS.
- J. C. Stiekema, Clin. Nephrol., 1986, 26, S3–S8 CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Y. Wang, S. J. Zhen, Y. Zhang, Y. F. Li and C. Z. Huang, J. Phys. Chem. C, 2011, 115, 12815–12821 CAS.
- X. Qi, K.-Y. Pu, X. Zhou, B. L. Hai Li, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663–669 CrossRef CAS.
- “Zeta potential of colloids in water and waste water”, ASTM Standard D4187-82, American Society for Testing and Materials, 1985..
- Y. Wang, Y. F. Li, J. Wang, Y. Sang and C. Z. Huang, Chem. Commun., 2010, 46, 1332–1334 RSC.
- Y. Wang, L. Q. Chen, Y. F. Li, X. J. Zhao, L. Peng and C. Z. Huang, Nanotechnology, 2010, 21, 305601 CrossRef.
- C. Xu and X. Wang, Small, 2009, 5, 2212–2217 CrossRef CAS.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390–412 CrossRef CAS.
- W. Hu, C. Peng, W. Luo, M. Lv, X. Li, D. Li, Q. Huang and C. Fan, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS.
- C. M. Jackson and Y. Nemerson, Annu. Rev. Biochem., 1980, 49, 765–811 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental details for the preparation of GO and hydrazine-rGO; Raman spectrum of graphite; AFM and SEM images of GO and heparin-rGO; XRD pattern of hydrazine-reduced rGO; Details for drug loading of heparin-rGO and corresponding absorption spectra; Data of anticoagulant activity investigation of graphene. See DOI: 10.1039/c2ra00841f/ |
| This journal is © The Royal Society of Chemistry 2012 |