Influence of nitrosyl coordination on the binding mode of quinaldate in selective ruthenium frameworks. Electronic structure and reactivity aspects†
Abhishek Dutta
Chowdhury
,
Prinaka
De
,
Shaikh M.
Mobin
and
Goutam Kumar
Lahiri
*
Department of Chemistry, IIT Bombay, Powai, Mumbai 400076, India. E-mail: lahiri@chem.iitb.ac.in; Fax: 91-022-2572-3480
First published on 29th February 2012
Abstract
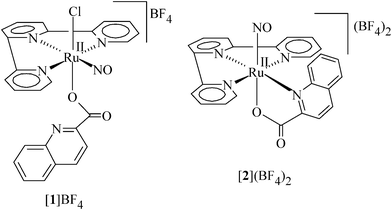
The nitrosyl complexes, [RuII(trpy)(L)(NO+)Cl]BF4, [1]BF4, and [RuII(trpy)(L)(NO+)](BF4)2, [2](BF4)2, (trpy = 2,2′:6′,2′′-terpyridine, L− = deprotonated form of unsymmetrical quinaldic acid) have been synthesized. Single crystal X-ray structures of [1]BF4 and [2](BF4)2 reveal that in the former L− binds to the ruthenium ion selectively in a monodentate fashion through the O− donor whereas the usual bidentate mode of L− (O−, N donors) has been retained in [2](BF4)2 with the same meridional configuration of trpy being seen in both. The Ru–NO group in [1]BF4 or [2](BF4)2, exhibits almost linear (sp-hybridized form of NO+) geometry. The difference in bonding mode of the unsymmetrical quinaldate in [1]BF4 and [2](BF4)2 has been reflected in their corresponding ν(NO)/ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) frequencies as well as in their NO based two-step reduction processes, {RuII–NO+} → {RuII–NO•} and {RuII–NO•}→{RuII–NO−}. The close to bent geometry (sp2-hybridized form of NO•) of the one-electron reduced 1 or [2]+ is been reflected in their DFT optimized structures. The spin density plot of the reduced species reveals that NO is the primary spin-bearing center with slight delocalization onto the metal ion which has been reflected in its radical EPR spectrum. [1]+ and [2]2+ undergo facile photorelease of NO with significantly different kNO (s−1) and t1/2 (s) values which eventually lead to the concomitant formation of the corresponding solvent species. The photoreleased NO• can be trapped as an Mb–NO adduct. The reduced species 1 selectively reacts with the molecular oxygen (O2) at pH ∼ 1 to yield the corresponding nitro species, [RuII(trpy)(L)(NO2)Cl]−.
O) frequencies as well as in their NO based two-step reduction processes, {RuII–NO+} → {RuII–NO•} and {RuII–NO•}→{RuII–NO−}. The close to bent geometry (sp2-hybridized form of NO•) of the one-electron reduced 1 or [2]+ is been reflected in their DFT optimized structures. The spin density plot of the reduced species reveals that NO is the primary spin-bearing center with slight delocalization onto the metal ion which has been reflected in its radical EPR spectrum. [1]+ and [2]2+ undergo facile photorelease of NO with significantly different kNO (s−1) and t1/2 (s) values which eventually lead to the concomitant formation of the corresponding solvent species. The photoreleased NO• can be trapped as an Mb–NO adduct. The reduced species 1 selectively reacts with the molecular oxygen (O2) at pH ∼ 1 to yield the corresponding nitro species, [RuII(trpy)(L)(NO2)Cl]−.
Introduction
The important role of nitric oxide (NO•) in various biological and physiological processes, such as blood pressure regulation, inflammatory response, and apoptosis has drawn renewed research interest on nitrosyl chemistry in recent years.1–7 NO• is generated in biological processes by nitric oxide synthase (NOS) during the oxidation of L-arginine to citrulline which plays important roles in neurotransmission, smooth muscle vasodilation and platelet disaggregation in mammals.6 The interaction of NO with the metal ions is important from the broader perspective of inorganic chemistry including in bioinorganic chemistry.4,5 For example, the reactions of NO• with oxygenated heme-protein have considerable biological significance.5 Though some organic nitrites, nitrates and S-nitrsothiols have medicinal application as vasodilators,2 they are not suitable for site specific NO• delivery processes. However, transition metal–nitrosyl complexes have shown potential application for site-specific NO•-delivery primarily due to photolabile nature of the metal–nitrosyl bond.8–10 Among them, ruthenium nitrosyl complexes have been emerged as a promising class of NO-donor due to their reduced activity towards oxygen and stability in water.8,9,11 Besides that, the non-innocent feature of NO facilitates its accessibility in three different redox states, strongly electrophilic NO+, neutral NO• and anionic NO−, in transition metal complexes depending on the electronic environment around the metal center which in turn makes the nitrosyl function as a versatile ligand in co-ordination chemistry.8,11 Further, ruthenium–nitrosyl complexes have shown considerable applications in pharmaceuticals, catalysis, molecular electronics and photochemical devices.12In this context the present article describes the various aspects of the coordinated nitrosyl function in the newly designed selective molecular frameworks of [RuII(trpy)(L)(NO)Cl]n, [1]n, and [RuII(trpy)(L)(NO)]n, [2]n, (trpy = 2,2′:6′,2′′-terpyridine, L− = deprotonated form of unsymmetrical quinaldic acid). Herein we report the synthesis and structural characterization of [1]BF4 and [2](BF4)2. Furthermore, the effect of structural diversity of the co-ligand, L− (monodentate versus bidentate) on the electronic structures of [1]n and [2]n with special reference to electrophilicity, redox potential and reactivity of the coordinated nitosyl function have been investigated by experimental studies and DFT calculations.
Results and discussion
Synthesis, characterization and structural aspects
The complexes with Enemark–Feltham notation13 of {Ru(NO)}6 in [RuII(trpy)(L)(NO+)Cl](BF4), [1]BF4, and [RuII(trpy)(L)(NO+)](BF4)2, [2](BF4)2, have been synthesized via the direct reaction of NOBF4 with the previously structurally characterized precursor complex [RuII(trpy)(L)Cl] (A)14 and the reaction of NOBF4 with the in situ generated [RuII(trpy)(L)(C2H5OH)]+, respectively, as shown in Scheme 1 (trpy = 2,2′:6′,2′′-terpyridine, L− = deprotonated form of unsymmetrical quinaldic acid, HL).15,16![The synthetic outline for [1]BF4 and [2](BF4)2.](/image/article/2012/RA/c2ra00953f/c2ra00953f-s1.gif) | ||
| Scheme 1 The synthetic outline for [1]BF4 and [2](BF4)2. | ||
[1]BF4 and [2](BF4)2 exhibit satisfactory microanalytical and mass spectral data (Fig. S1†) and show 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 molar conductivities in acetonitrile solution, respectively (see Experimental Section).
:1 and 1:2 molar conductivities in acetonitrile solution, respectively (see Experimental Section).
The formation of [1]BF4 and [2](BF4)2 have been authenticated by their single crystal X-ray structures (Fig. 1 and Table 1). The five-membered chelate ring of L− (through the O1− and N1 donors of L−) in the precursor A has been retained in [2](BF4)2 along with the usual meridional configuration of trpy.14 However, the direct nitrosylation of A (Scheme 1) surprisingly leads to the concomitant transformation of the bidentate (O−, N donors) mode of L− to the monodentate (O− donor) in [1]BF4 retaining the same meridional configuration of trpy.
![ORTEP diagrams of (a) [1]BF4·5H2O and (b) [2](BF4)2. Thermal ellipsoids are drawn at 50% probability. The solvents of crystallization, counter anions and hydrogen atoms are omitted for clarity.](/image/article/2012/RA/c2ra00953f/c2ra00953f-f1.gif) | ||
| Fig. 1 ORTEP diagrams of (a) [1]BF4·5H2O and (b) [2](BF4)2. Thermal ellipsoids are drawn at 50% probability. The solvents of crystallization, counter anions and hydrogen atoms are omitted for clarity. | ||
| [1]BF4·5H2O | [2](BF4)2 | |
|---|---|---|
| Empirical formula | C25H17B1Cl1F4N5O8Ru | C25H17B2F8N5O3Ru |
| Mr | 738.77 | 710.13 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 8.5160(3) | 8.4478(2) |
| b/Å | 10.1858(4) | 8.9532(2) |
| c/Å | 16.7736(6) | 17.6431(4) |
| α (°) | 92.631(3) | 94.079(2) |
| β (°) | 102.713(3) | 91.367(2) |
| γ (°) | 93.147(3) | 98.405(2) |
| V/Å3 | 1414.66(9) | 1315.96(5) |
| Z | 2 | 2 |
| μ/mm−1 | 0.733 | 0.693 |
| T/K | 150(2) K | 120(2) |
| D c/g cm−3 | 1.734 | 1.792 |
| F(000) | 736 | 704 |
| 2θ range (°) | 6.54 to 50 | 6.58 to 50 |
| data/restraints/parameters | 4952/0/434 | 4622/0/397 |
| R 1, wR2 [I > 2σ] | 0.0698, 0.2092 | 0.0396, 0.1046 |
| R 1, wR2 (all data) | 0.0795, 0.2136 | 0.0435, 0.1067 |
| GOF | 1.136 | 1.061 |
| largest diff. peak, hole/e Å−3 | 2.399 and −0.755 | 1.307 and −0.889 |
The bond distances and bond angles in [1]BF4 and [2](BF4)2 (Table 2) are in good agreement with the reported data of analogous complexes.8,9,11 The geometrical constraint due to the meridional mode of trpy has been reflected in the appreciably smaller trans angles involving the trpy ligand, N2–Ru–N4 of 156.5(3)° and 159.35(12)° in [1]BF4 and [2](BF4)2, respectively. The central Ru–N3(trpy) bond lengths of 2.015(6) Å in [1]BF4 and 1.981(3) Å in [2](BF4)2 are significantly shorter than the corresponding distances involving the terminal pyridine rings of trpy, Ru–N2(trpy), 2.083(6) Å and 2.075(3) Å, and Ru–N4(trpy), 2.079(6) Å and 2.079(3) Å, respectively. The central Ru–N3(trpy) distance in [1]BF4 is 0.034 Å longer than that in [2](BF4)2 due to the effect of the strongly π-accepting NO+trans to the Ru–N3(trpy). The Ru–O−(monodentate L−) bond distance in [1]BF4 (2.044(5) Å) is appreciably longer relative to Ru–O−(chelated L−) in [2](BF4)2 (1.990(2) Å) due to the effect of oppositely directed σ/π-donor Cl−versus the strongly π-accepting NO+trans to the Ru–O−(L−) bond. The impact of varying coordination situations in the complexes has been reflected in their trans angles: O1–Ru–Cl, 171.85(15)° and N3–Ru–N5, 175.6(3)° in [1]BF4versus O1–Ru–N5, 176.43(12)° and N(3)–Ru–N(1), 163.12(12)° in [2](BF4)2. The almost linear mode of Ru–N5–O3 (NO), 173.0(8)° in [1]BF4 and 175.9(3)° [2](BF4)2 as well as the triple bond feature of N5![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O3 (NO) (1.141(9) Å in [1]BF4 and 1.130(4) Å in [2](BF4)2) establish the sp-hybridized state of the nitrogen atom of the NO+ group as expected from the coordinated π-accepting nitrosonium ion. However, the presence of the π-accepting trpy ligand (N3) trans to Ru–N5–O3 in [1]BF4 makes the Ru–N–O angle (173.0(8)°) relatively more tilted compared to that in [2](BF4)2 (175.9(3)°) where the Ru–N–O group is trans to the σ-donating O− of the chelated L−. The pendant quinaldate ring in [1]BF4 is slightly tilted (about 96°) with respect to the equatorial plane. The Ru–O1–C1 angle of 118.9(5)° implies that the bound oxygen (O1) is close to the sp2-hybridized state which in turn introduces a resonating feature of the carboxylate group (O1–C1, 1.276(9) Å and C1–O2, 1.242(10) Å) in [1]BF4 (Table 2).
O3 (NO) (1.141(9) Å in [1]BF4 and 1.130(4) Å in [2](BF4)2) establish the sp-hybridized state of the nitrogen atom of the NO+ group as expected from the coordinated π-accepting nitrosonium ion. However, the presence of the π-accepting trpy ligand (N3) trans to Ru–N5–O3 in [1]BF4 makes the Ru–N–O angle (173.0(8)°) relatively more tilted compared to that in [2](BF4)2 (175.9(3)°) where the Ru–N–O group is trans to the σ-donating O− of the chelated L−. The pendant quinaldate ring in [1]BF4 is slightly tilted (about 96°) with respect to the equatorial plane. The Ru–O1–C1 angle of 118.9(5)° implies that the bound oxygen (O1) is close to the sp2-hybridized state which in turn introduces a resonating feature of the carboxylate group (O1–C1, 1.276(9) Å and C1–O2, 1.242(10) Å) in [1]BF4 (Table 2).
| Bond distance (Å)/Bond angle (°) | [1]+ | [2]2+ | ||
|---|---|---|---|---|
| X-Ray | DFT | X-Ray | DFT | |
| Ru–N1 | — | — | 2.116(3) | 2.19 |
| Ru–N2 | 2.083(6) | 2.11 | 2.075(3) | 2.14 |
| Ru–N3 | 2.015(6) | 2.03 | 1.981(3) | 2.02 |
| Ru–N4 | 2.079(6) | 2.11 | 2.079(3) | 2.13 |
| Ru–N5 | 1.754(7) | 1.79 | 1.758(3) | 1.80 |
| Ru–O1 | 2.044(5) | 2.05 | 1.990(2) | 1.97 |
| Ru–Cl | 2.361(2) | 2.44 | — | — |
| O(1)–C1 | 1.276(9) | 1.30 | 1.317(4) | 1.34 |
| O(2)–C1 | 1.242(10) | 1.24 | 1.202(5) | 1.21 |
| N(5)–O3 | 1.141(9) | 1.14 | 1.130(4) | 1.15 |
| N1–Ru–N3 | — | — | 163.12(12) | 160.82 |
| N2–Ru–N4 | 156.5(3) | 156.32 | 159.35(12) | 158.22 |
| O1–Ru–N5 | 98.3(3) | 97.80 | 176.43(12) | 175.03 |
| N3–Ru–N5 | 175.6(3) | 175.95 | 94.83(13) | 94.98 |
| Cl–Ru–O1 | 171.85(15) | 171.59 | — | — |
| N5–Ru–N2 | 100.1(3) | 100.80 | 96.49(13) | 90.86 |
| N5–Ru–N3 | 175.6(3) | 175.95 | 94.83(13) | 94.98 |
| N5–Ru–N4 | 103.4(3) | 102.88 | 91.29(13) | 95.83 |
| N5–Ru–N1 | — | — | 101.82(12) | 104.18 |
| N1–Ru–N4 | — | — | 96.52(11) | 99.17 |
| N3–Ru–O1 | 85.8(2) | 86.11 | 84.26(11) | 81.61 |
| Ru–N5–O3 | 173.0(8) | 170.25 | 175.9(3) | 174.02 |
| Ru-1-C1 | 118.9(5) | 117.21 | 116.6(2) | 119.15 |
The DFT calculated bond parameters (Table 2) based on the optimized structures of [1]+ and [2]2+ (Fig. S2†) are in general agreement with the X-ray data.
The corresponding one-electron reduced species with {Ru(NO)}7 configuration in [RuII(trpy)(L)(NO•)Cl] (1) and [RuII(trpy)(L)(NO•)]+ ([2]+) can be generated electrochemically.8,11 The DFT calculated structural parameters of [1]+/1 and [2]2+/[2]+ (Fig. 2 and Table S1†) reveal similar structural differences between the two sets of complexes based on the electronic environment around the {Ru–NO} groups. The σ-donating ability of the carboxylate oxygen (O1) of L− makes the Ru–N5–O3 angle relatively more bent in [2]+ (139.6°) than in 1 (141.85°) where the Ru–NO group is situated trans to the π-accepting trpy (N3) ligand (Fig. 3). The difference in calculated Ru–N5–O3 angles between [2]2+ and [2]+ of ∼35° is reasonably larger than that between [1]+ and 1 (∼29°) implying greater electron density on N(5) in [2]+ as has also been revealed by the NBO studies (Table 3). The double bond feature of N5–O3 (NO), 1.176 Å and 1.18 Å in 1 and [2]+, respectively, suggests the sp2 character of the nitrogen atom. The lengthening of the Ru–N5 bond upon NO based reduction, 0.14 Å for [1]+/1 and 0.12 Å for [2]2+/[2]+ due to increasing electron–electron repulsion upon addition of an electron is in agreement with the established concept of labilization of the Ru–NO bond on reduction.11h
![The DFT optimized geometry of (a) 1 and (b) [2]+. The hydrogen atoms are omitted for clarity.](/image/article/2012/RA/c2ra00953f/c2ra00953f-f2.gif) | ||
| Fig. 2 The DFT optimized geometry of (a) 1 and (b) [2]+. The hydrogen atoms are omitted for clarity. | ||
![Schematic representation of 1 and [2]+.](/image/article/2012/RA/c2ra00953f/c2ra00953f-f3.gif) | ||
| Fig. 3 Schematic representation of 1 and [2]+. | ||
| Total atomic charge | Natural charge | |||||||
|---|---|---|---|---|---|---|---|---|
| Ru | N(5) | O(3) | Cl | Ru | N(5) | O(3) | Cl | |
| [1]+ | 0.733 | 0.135 | −0.068 | −0.226 | 0.737 | 0.383 | −0.065 | −0.491 |
| [1] | 0.617 | 0.026 | −0.182 | −0.349 | 0.635 | 0.184 | −0.194 | −0.576 |
| [2]2+ | 1.013 | 0.054 | −0.070 | — | 0.802 | 0.317 | −0.072 | — |
| [2]+ | 0.840 | −0.022 | −0.189 | — | 0.687 | 0.139 | −0.196 | — |
Spectral and redox aspects
The 1H NMR spectra of [1]+ and [2]2+ in (CD3)2SO exhibit the calculated number of seventeen partially overlapping aromatic proton resonances in each case (Fig. S3†). The extent of overlap of the proton signals is appreciably larger in [2]2+ as compared to [1]+. The maximum downfield shifted signals in [2]2+ and [1]+ appear at 10.2 ppm and 9.2 ppm, respectively. This can be attributed to the proton trans to the quinoline nitrogen N(1) of the chelated L− opposite to the central pyridine ring of the π-acceptor trpy in [2]2+ whereas the same atom in the monodentate quinoline ring in [1]+ exists away from the coordination sphere.The MOs of [1]+ and [2]2+ (Tables S2 and S4†) predict that in both cases the HOMOs are primarily composed of L− based orbitals. The LUMO and LUMO + 1 are however dominated by NO based orbitals which in effect make them susceptible towards reduction (Tables S2 and S4†). Accordingly, [1]+ and [2]2+ exhibit two successive reductions, E°298,V (ΔEp, mV) at 0.31(70), −0.34(80) and 0.05(60), −0.64(90) in CH3CN versus SCE corresponding to the {RuII–NO+} → {RuII–NO•} (I) and {RuII–NO•} → {RuII–NO−} (II) couples, respectively (Fig. 4).8,11 The selective trans orientation of two π-accepting groups, trpy and NO+ in [1]+ makes the reduction processes relatively easier than that in [2]2+ where the NO+ is trans to the electron rich O− donor of L−. This has also been reflected in the greater natural charge on N(5) (NO) in [1]+ than [2]2+ (Table 3). The significant contribution of trpy based orbitals in higher UMOs results in trpy based quasi-reversible reductions at further negative potentials, E°298,V (ΔEp, mV) of −0.84(100), −1.65(110) and −0.7(110), −1.15(110) for [1]+ and [2]2+, respectively.8a–c,11a,h–m,14
![Cyclic voltammograms of (a) [1]+ and (b) [2]2+ in CH3CN/0.1 M [Et4N](ClO4) versus SCE, scan rate:100 mV s−1.](/image/article/2012/RA/c2ra00953f/c2ra00953f-f4.gif) | ||
| Fig. 4 Cyclic voltammograms of (a) [1]+ and (b) [2]2+ in CH3CN/0.1 M [Et4N](ClO4) versus SCE, scan rate:100 mV s−1. | ||
[1]+ and [2]2+ exhibit moderately intense transitions in the near-UV region, 326 nm and 348 nm, respectively, followed by several intense higher energy intraligand transitions in the UV region in CH3CN (Fig. 5).8,11 The bands in the near-UV region are assigned on the basis of the TD-DFT calculations on the optimized structures of [1]+ and [2]2+ as the RuII(dπ)/L(π) → NO+(π*) transition (Tables S5 and S6†). The slight difference in electronic spectra in the complexes can be attributed to the mixing of chloride ion orbitals in the HOMOs of [1]+ and the denticity of L−. Upon one-electron reduction to [1] and [2]+ the near UV region bands shift to the lower energy region at 556 nm and 493 nm, respectively, which are assigned to the RuII(dπ)/NO•(π) → π*(trpy) and RuII(dπ)/NO•(π) → L(π*) transitions, respectively, based on the TD-DFT calculations.
![Electronic spectra in CH3CN of (a) [1]+ (black), 1 (red) and (b) [2]2+ (black), [2]+ (red).](/image/article/2012/RA/c2ra00953f/c2ra00953f-f5.gif) | ||
| Fig. 5 Electronic spectra in CH3CN of (a) [1]+ (black), 1 (red) and (b) [2]2+ (black), [2]+ (red). | ||
The ν(CO) frequency of the coordinated L− in the precursor [(RuII(trpy)(L)(Cl)] (A) at 1635 cm−1.14 has been appreciably shifted to 1667 cm−1 and 1696 cm−1 in [1]BF4 and [2](BF4)2, respectively (Fig. S4†) due to their different electronic environments. The characteristic vibrations of the BF4− counter anion appear near 1600 cm−1 and 1080 cm−1. The reasonably high ν(NO) frequencies of [1]BF4 and [2](BF4)2 of 1895 cm−1 (DFT: 1906 cm−1) and 1926 cm−1 (DFT: 1939 cm−1), respectively, imply their moderately electrophilic character. Upon one-electron reduction to 1 and [2]+, the NO bands at 1895 cm−1 and 1926 cm−1 disappear. However, the ν(NO•) frequency of the reduced state in the expected SWIR region of 1600–1700 cm−1 did not resolve properly due to the presence of other characteristic vibrations of the CO of coordinated L−, BF4− counter anion as well as aryl ring vibrations.
The EPR spectrum of the representative one-electron reduced paramagnetic [2]+ in frozen CH3CN/0.1 M Bu4N(PF6) solution yields g-components at 2.012 (g1), 1.988 (g2) and 1.869 (g3) (Fig. S5†) with a g-anisotropy (g1 − g3) of 0.143, which is about twice a large as that observed for the related {Fe(NO)}7 species due to ζ(Ru) ≈ 2ζ(Fe) (ζ = spin–orbit coupling constant).10b,11h However, the value of <g> = [1/3(g12 + g22 + g32)]1/2 = 1.957 implies that the spin is primarily localized on the N(5) of NO.17,18 The overall negative shift of the g value (Δg = 45.0 ppt) with respect to the free electron value of 2.0023 arises due to the admixture of higher excited states with nonzero angular momentum (eqn (1)).18 The radical EPR feature of [2]+ (Fig. S5†) has been reflected in the significant bending of Ru–N(5)–O(3) angle (139.69°) and lengthening of the N(5)–O(3) bond (1.181 Å).
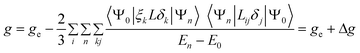 | (1) |
ξ: spin orbit coupling constant
L: angular momentum operator
E 0: energy of the SOMO
The SOMO of [2]+ indicates σ-donation from the NO(π*) orbital to the Ru(dz2) center which reveals {RuII–NO•} as the predominant oxidation state formulation along with considerable mixing of the {RuI–NO+} state due to the presence of 23% metal contribution. The presence of ∼10% spin on the metal in spin density analysis also reveals that spin is mainly concentrated on NO (Fig. 6). Moreover, σ-interaction between Ru and NO results in spin density at the axial position (Fig. 6) which leads to a significant hyperfine splitting in the EPR spectrum due to the N(5) of NO.
![Mulliken atomic spin density plot for [2]+ (N(5): 0.514, O(3): 0.329, Ru: 0.096).](/image/article/2012/RA/c2ra00953f/c2ra00953f-f6.gif) | ||
| Fig. 6 Mulliken atomic spin density plot for [2]+ (N(5): 0.514, O(3): 0.329, Ru: 0.096). | ||
Electronic features: theoretical insights
Molecular orbital analysis reveals that the LUMOs of [1]+ and [2]2+ originate from the interaction of the π*-orbital of NO+ with the metal's t2g(dxy) and eg(dz2) orbitals, respectively, (Tables S2 and S4†).10b,11c–h,17 This is attributed to stronger back-bonding in [1]+ between the metal t2g(dπ) orbital and the π*-orbital of trpy in the filled MOs. The presence of 10–25% metal contribution in the LUMOs lowers the energy of the metal t2g(dπ) orbitals and thus the HOMOs are mainly composed of the orbitals of the co-ligands (trpy, L or Cl).10b The significant metal contribution (∼58%) in [2]2+ has been predicted in the HOMO − 5 state whereas around 40% metal contribution in [1]+ has been detected in HOMO − 11 which also provides evidence for the lesser extent of back-bonding in [2]2+. The presence of a σ/π-donating chloride ion plays a crucial role for the stronger back-bonding in [1]+. This is reflected in a greater natural charge on the metal ion in [2]2+ than [1]+ (Table 3). The observed more linearity of the {Ru–N(5)–O(3)} bond in [2]2+ (175.9(3)°) as compared to [1]+ (171.0(8)°) suggests relatively larger degeneracy of the π*-orbitals (LUMO and LUMO + 1) in [2]2+.The addition of one-electron to the π*-orbital of {RuNO}6 in [1]+ or [2]2+ leads to the formation of a {RuNO}7 species, 1 or [2]+ and hence lifts the said degeneracy of the π*-orbitals (LUMO and LUMO + 1) (Tables S3–S4†).10b,17a Such a splitting can be viewed as a Jahn–Teller splitting due to the lowering of symmetry and the spin–orbit interaction which is reflected in the bending mode of Ru–NO with Ru–N(5)–O(3) angles of 141.85° and 139.69 in 1 and [2]+, respectively, as revealed in their DFT optimized structures. The SOMO of 1 or [2]+ is primarily composed of the 2p(π)-atomic orbitals of N5 and O3 (π*(NO•)) along with a lesser extent of 4dxy(RuII) orbital of 1 and dz2(RuII) orbital of [2]+. The appreciable extent of metal contribution in the SOMO suggests {RuII–NO•} as the predominant oxidation state along with partial mixing of the {RuI–NO+} state.11c–i,l The SOMO of 1 suggests the presence of a 4dxy(RuII) → pπ*(NO•) back-bonding interaction while the SOMO of [2]+ reveals the σ-charge donation from the low-lying singly-occupied pπ*(NO•) to the metal eg-orbitals. This can be attributed to the different coordination environment of the NO function in the complexes: NO is trans to the π-accepting trpy ligand in the equatorial plane in [1]+ whereas in [2]2+ the NO is trans to the σ-donating O− of L− in an axial orientation.
The approximate bent geometry of {Ru–NO}7 in 1 and [2]+ introduces the possibility of a pair of conformational isomers based on the dihedral angle (θ) of Ltrans–Ru–N5–O3, with the idealized eclipsed and staggered angles of θ = 0° and θ = 45°, respectively.11b–d,h The geometry optimization at the G03/(U)B3LYP level reveals that the eclipsed conformation is more stable by 0.01 eV in [2]+. However, neither eclipsed nor staggered conformation is found to be perfectly suitable for 1 where the dihedral angle (θ) of Ltrans(N3)–Ru–N5–O3 is 28.3° (Fig. S6†). Such a pseudo-staggered (θ = 28.3°) conformation in 1 is more stable by 0.05 eV and 0.12 eV with respect to the eclipsed (θ = 0°) and staggered (θ = 45°) forms, respectively. Another theoretically possible staggered conformation with the dihedral angle of θ = −45° is destabilized under the same level of DFT presumably due to the repulsive interaction between O3 and O2. The stereoelectronic effect of the chloride and O2 from bulky L in 1 collectively prevent it exhibiting either the eclipsed (θ = 0°) or staggered (θ = 45°) conformation, leading to an energetically favored pseudo-staggered (θ = 28.3°) conformation.
Photo-lability of the {Ru–NO} bond
The facile photo-labilization of the {RuII–NO•} bond maintaining the integrity of the remaining part of the molecule is believed to be significant particularly from the perspective of the biochemically desired target oriented NO• delivery process.8,9 In this context both the nitrosyl complexes, [RuII(trpy)(L)(NO+)Cl]+, [1]+, and [RuII(trpy)(L)(NO+)]2+, [2]2+, are found to undergo the facile photocleavage of the {Ru–NO} bond under the exposure of light in CH3CN as evidenced by their spectroscopic signatures (Fig. 7).19 The photolabilization of the {Ru–NO} bond is accompanied by the formation of the corresponding solvent bound ruthenium(II)-photoadducts, [RuII(trpy)(L)(Cl)(CH3CN)] and [RuII(trpy)(L))(CH3CN)]+, respectively, and the transformation proceeds through several isosbestic points (Fig. 7). The formation of an Mb–NO adduct on passing the liberated “NO” through the aqueous solution of reduced Mb under deoxygenated conditions has been evidenced by its characteristic absorption band at λmax = 420 nm (Fig. S7†). The estimated first-order rate constant (k/s−1) and t1/2/s values of the photocleavage process are 2.6 × 10−1, 2.66 and 2.9 × 10−2, 23.8 for [1]+ and [2]2+, respectively. The ten-fold faster photolability of the {RuII–NO•} bond in [1]+ as compared to [2]2+ can be rationalized based on their structural differences including the trans influence of co-ligands. The cleavage of the {RuII–NO+} bond via the photo-irradiation process is known to proceed through the formation of the intermediate excited S = 1 state in {RuIII–NO•}* as shown below.8f![Time evolution of the electronic spectra of (a) [1]+, concentration: 0.57 × 10−4 M in CH3CN, time intervals: 2 s, and (b) [2]2+, concentration: 0.21 × 10−5 M in CH3CN, time intervals: 5 s under the exposure of light (Xe lamp, 350 W). Insets show the absorbance versus time plots at (a) 326 nm and (b) 482 nm corresponding to the solvent species in each case.](/image/article/2012/RA/c2ra00953f/c2ra00953f-f7.gif) | ||
| Fig. 7 Time evolution of the electronic spectra of (a) [1]+, concentration: 0.57 × 10−4 M in CH3CN, time intervals: 2 s, and (b) [2]2+, concentration: 0.21 × 10−5 M in CH3CN, time intervals: 5 s under the exposure of light (Xe lamp, 350 W). Insets show the absorbance versus time plots at (a) 326 nm and (b) 482 nm corresponding to the solvent species in each case. | ||
[RuII–NO+] + hν → [RuIII–NO•]* → [RuII–solvent] + NO•
The formation of a solvated Ru(II) species instead of Ru(III)–solvate as a photoproduct has been attributed to the absence of the otherwise expected EPR signal of Ru(III) as has also been established earlier.8 The electronic effect of π-accepting ligands certainly facilitates the formation of {RuII–CH3CN} in the ground state of the photo-product, as has also been established recently by us.8g It should be noted that the selective photocleavage of the M–NO bond, maintaining the integrity of the rest of the molecule is biologically significant.9 The competition between the pπ-orbitals of NO and N3(trpy) for the same dπ-orbital of the metal ion (observed in HOMO − 4) makes the {Ru–NO} bond in [1]+ more photolabile. On the other hand, the greater extent of dπ(Ru) → pπ(NO) back-bonding as well as trans-influence of the σ-donating carboxylate group (O1 of L−) are the likely factors for the relatively slower photo-dissociation process in [2]2+.
Reactivity of {Ru–NO} towards molecular oxygen: dioxygenase activity
The reaction of NO with the oxygenated heme-proteins has immense biological importance.5 Though nitrogen monoxide plays crucial roles in various physiological processes, overproduction of NO can also lead to several toxicological processes, such as cell death and DNA damage, primarily due to the formation of highly reactive peroxynitrite (−OONO).20 Nitric oxide dioxygenase (NODs) catalyzes the reaction of NO with dioxygen to yield the environmentally and physiologically benign nitrate anion (NO3−) in mammals.21 Highly reactive peroxynitrite (−OONO or oxoperoxonitrate) is known to be the powerful oxidant/nitrating agent in nitric oxide biochemistry and has an influential role in stress injury.21 The selective conversion of peroxynitrite (−OONO) to biologically benign nitrate takes place in oxygenated heme-proteins to control the overproduction of NO•22,23but in synthetic model systems, peroxynitrite usually transforms to nitrite.24a–b,d
Although the reduced [2]+ remains inactive towards molecular oxygen (O2), 1 (in situ generated via the addition of N2H4 in the CH3CN solution of [1]+ or [2]2+) is found to activate molecular oxygen at 298 K. Upon bubbling dioxygen (O2) in an acetonitrile solution of 1 at pH ∼ 1, the intensity of the peaks at 525 nm and 376 nm corresponding to 1 periodically decreases with the concomitant growth of a new peak at 476 nm (Fig. S8†). The transformation proceeds through several isosbestic points at 501 nm, 399 nm and 355 nm, implying a clean process with the selective involvement of two species, [RuII(trpy)(L)(NO•)Cl] (1) and [RuII(trpy)(L)(NO2)Cl]− ([1a]−). The formation of [1a]− is evidenced from its ESI-MS(+) peak at 554.88 corresponding to {([1a]–Cl) + H+} (calcd. mass: 554.03) (Scheme 2 and Fig. S9†). The transformation of 1 to [1a]− is likely to occur via the intermediate transient peroxynitrite species {Ru–(OONO)} ([1a′]−). Consequently, the oxygenation reaction of 1 in the presence of tyrosine-mimic 2,4-di-tert-butylphenol at pH ∼ 1 results in the nitrated product, 2,4-di-tert-butyl-6-nitrophenol (NO2–DTBP) and oxidative coupling product, 2,2′-dihydroxy-3,3′,5,5′-tetra-tert-butyl-1,1′-biphenyl as depicted in Scheme 2.24
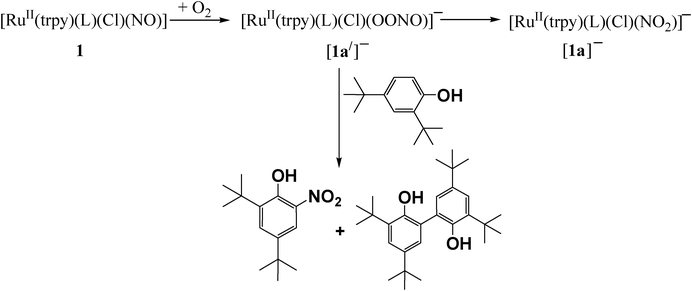 | ||
| Scheme 2 Dioxygenase activity of 1 at pH ∼ 1. | ||
Conclusions
The present article highlights the following points:- The unprecedented hemilabile feature of the chelated quinaldate ligand (L−) in the presence of NO+ in the molecular framework of [1]+ comprising of selective co-ligands, π-acepting trpy and σ/π-donating chloride.
- The electronic aspects of the coordinated nitrosyl function in [1]BF4 and [2](BF4)2 differ appreciably based on their specific structural features as evidenced by their ν(NO) frequencies and E°(RuII–NO+/RuII–NO•) potentials.
- The built in electronic structural differences in [1]+ and [2]2+ have further been reflected in the rate of light induced {RuII–NO+} bond cleavage.
- Reduced 1 has been selectively transformed to the corresponding {RuII–NO2} species through contact with molecular oxygen, although the corresponding [2]+ failed to exhibit any such activity with O2.
Experimental section
Materials
The precursor complexes Ru(trpy)Cl325a and [RuII(trpy)(L)(Cl)] 14 (trpy = 2,2′:6′,2′′-terpyridine) were prepared according to literature procedures. The ligand quinaldic acid (HL) and other reagents and chemicals were obtained from Aldrich and used as received. For spectroscopic and electrochemical studies HPLC-grade solvents were used.Physical measurements
1H NMR spectra were recorded in (CD3)2SO on a 400 MHz Bruker spectrometer. Chemical shift data are quoted as δ in ppm and as s, d, dd, t, q and m representing singlet, doublet, doublet of doublet, triplet, quartet, sextet and multiplet peaks, respectively. IR and UV-vis spectra were recorded using Thermo Nicolet 320 and Perkin Elmer Lambda 950 spectrophotometers, respectively. ESI-mass spectra were recorded using micromass Q-TOF. Cyclic voltammetric studies were carried out using a PAR model 273A electrochemistry system. Platinum wire working and auxiliary electrodes and an aqueous SCE were used in a three-electrode configuration. The supporting electrolyte was 0.1 mol dm−3 [NEt4](ClO4), and the solute concentration was ∼10−3 mol dm−3. The half-wave potential E°298 was set equal to 0.5(Epa + Epc), where Epa and Epc are the anodic and cathodic cyclic voltammetric peak potentials, respectively. Elemental analyses were carried out on Perkin-Elmer 240C elemental analyzer. The 2,4-di-tert-butyl phenol (DTBP) reactions were monitored by gas chromatographic technique with a FID detector (Shimadzu GC-2014 gas chromatograph) as well as GCMS (Hewlett-Packard GCD-HP1800A). The EPR measurement was made in a two-electrode capillary tube with a X-band Bruker system ESP300.25b:1 acetonitrile–benzene solution. Yield: 45 mg (75%). Anal. calcd. for C25H17N5ClO3BF4Ru (Mol. wt.: 659.01): C, 45.52; H, 2.60; N, 10.62. Found: C, 45.35; H, 2.58; N, 10.53%. Molar conductivity (ΛM [Ω−1 cm2 M−1], CH3CN): 105. ESI(+)-MS (m/z, CH3CN): 572.05 ([1]+), 543.04 ([1–NO + H]+), 537.08 ([1–NO–Cl]+). 1H NMR (400 MHz, (CD3)2SO): δ (ppm) (J/Hz): 9.25 (1H, d, 8.4), 9.09 (2H, d, 8.4), 9.02 (2H, d, 8.0), 8.97 (1H, d, 7.2), 8.70 (1H, d, 8.8), 8.67 (3H, m), 8.34 (2H, m), 8.15 (1H, t, 7.84, 7.20), 8.02 (2H, d, 7.3), 7.79 (2H, m). IR (KBr) ν(BF4−): 1083, 1601; ν(NO+):1895; ν(CO): 1667 cm−1. λ/nm (CH3CN) (ε/dm3 mol−1 cm−1): 455(sh), 349(10380), 326(15120), 315(14120), 283(16540).
:1 benzene–acetonitrile. Yield: 26 mg (55%). Anal. calcd. for C25H17N5O3B2F8Ru (Mol. wt. 711.04): C, 42.19; H, 2.41; N, 9.85. Found: C, 42.25; H, 2.45; N, 9.89%. Molar conductivity (ΛM [Ω−1 cm2 M−1], CH3CN): 190. ESI(+)-MS (m/z, CH3CN): 624.31 ([2](BF4)2–BF4]+). 1H NMR (400 MHz, (CD3)2SO): δ (ppm) (J/Hz): 10.25 (1H, d, 8.5), 9.18 (2H, m), 8.92 (5H, m), 8.49 (2H, m), 8.28 (1H, d, 8.4), 7.94 (3H, m), 7.75 (1H, m), 7.61 (2H, m). IR (KBr), ν(BF4−): 1083, 1599; ν(NO+):1927; ν(CO): 1696 cm−1. λ/nm (CH3CN) (ε/dm3 mol−1 cm−1): 346(20920), 325(20360), 289(24380), 279(26770).
Trapping of photoreleased “NO” by myoglobin
3.0 cm3 acetonitrile solution of the nitroso species ([1]+ or [2]2+) was initially taken in a quartz cuvette of optical path length of 1 cm. The cuvette was sealed with a rubber septum and the solution was deoxygenated by purging nitrogen gas. The photolyis was carried out for 10 min using a Xe 350 W lamp. The photoreleased free “NO” was allowed to pass through the reduced myoglobin solution in water using a cannula and the UV-vis. spectrum was recorded.Dioxygenase activity
The conversion process of [RuII(trpy)(L)(NO•)Cl] (1) → [RuII(trpy)(L)(NO2)Cl]− ([1a]−) in acetonitrile at pH ∼ 1 and in presence of bubbling O2 was monitored by following the increase in absorbance of the new band at 476 nm corresponding to λmax of the nitro species [1a]− till the changes of intensity completely levelled off. The above experiment was then repeated in presence of 2,4-di-tert-butylphenol (DTBP) at 298 K and the formation of corresponding products, 2,4-di-tert-butyl-6-nitrophenol (NO2-DTBP) and oxidative coupling product, 2,2′-dihydroxy-3,3′,5,5′-tetra-tert-butyl-1,1′-biphenyl were confirmed by GC and GCMS.Crystallography
Single crystals of [1]BF4 and [2](BF4)2 were grown by slow evaporation of their 1:1 acetonitrile–benzene solutions. Single crystal X-ray diffraction data were collected using an OXFORD XCALIBUR-S CCD single crystal X-ray diffractometer at 150 K. The structures were solved and refined by full-matrix least-squares techniques on F2 using the SHELX-97 program.26 All data were corrected for Lorentz polarization and absorption effects, and the non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included in the refinement process as per the riding model. The complex [1]BF4 was crystallized with five water molecules. CCDC-838835 and CCDC-838836 contain the supplementary crystallographic data for [1]+ and [2]2+, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
Computational details
Full geometry optimizations were carried out at the (R)B3LYP and (U)B3LYP levels27,28 using density functional theory method with Gaussian 03 (revision C.02).29 All elements except ruthenium were assigned the 6-31G(d) basis set during geometry optimizations and 6-31G(d,p) during single point and property calculations. The LanL2DZ basis set with effective core potential was employed for the ruthenium atom.30,31 Vertical electronic excitations based on (R)/(U)-B3LYP optimized geometries were computed for the time-dependent density functional theory (TD-DFT)32 in acetonitrile using the Polarizable continuum model (PCM) of Tomasi and co-workers,33 specifically, the conductor like PCM (CPCM) in conjugation with the united atom topological model (UAO radii, implemented in Gaussian 03) was applied.33–35 GaussSum was used to calculate the fractional contributions of various groups to each molecular orbital.36 No symmetry constraints were imposed during structural optimizations, and the nature of the optimized structures and energy minima were defined by subsequent frequency calculations. Natural bond orbital analyses were performed using the NBO 3.1 module of Gaussian 03 on optimized geometry.37 All the calculated structures were visualized with ChemCraft.38Acknowledgements
The financial support received from Department of Science and Technology (DST), New Delhi and Council of Scientific and Industrial Research (CSIR), New Delhi (fellowship to A.D.C.) and University Grant Commission (UGC) (fellowship to P.D.), New Delhi is gratefully acknowledged. X-Ray and GCMS studies were carried out at the National Single-Crystal X-ray Diffraction Facility and Sophisticated Analytical Instrumentation Facilities (SAIF), IIT Bombay, respectively. Computational facilities from the Department of Chemistry, IIT Bombay are gratefully acknowledged. We thank Mr. Thomas Scherer, Institut für Anorganische Chemie, Universität Stuttgart for EPR measurement.References
- (a) L. J. Ignarro, Hypertension, 1990, 16, 477 CAS; (b) S. Kalsner, ed., Nitric Oxide Free Radicals in Peripheral Neurotransmission, Birkhauser: Boston, 2000 Search PubMed; (c) G. Y. Ko and F. C. Fang, Nitric Oxide and Infection, KluwerAcademic/Plenum Publishers, New York, 1999 Search PubMed; (d) J. Lincoln and G. Burnstock, Nitric Oxide in Health and Disease, Cambridge University Press, New York, 1997 Search PubMed; (e) F. T. Bonner and G. Stedman, in Methods in Nitric Oxide Research, ed. M. Feelisch and J. S. Stamler, Wiley: Chichester, U.K., 1996 Search PubMed; (f) P. G. Wang, T. B. Cai and N. Taniguchi, Nitric Oxide Donors for Pharmaceutical and Biological Applications, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (g) F. Bottomley, in Reactions of Coordinated Ligands, ed. P. S. Braterman, Plenum, New York, 1985, 2, 115 Search PubMed; (h) F. Bottomley, Coord. Chem. Rev., 1978, 26, 7 CrossRef CAS ; Acc. Chem. Res., 1978, 11, 158.
- (a) A. Calver, J. Collier and P. Vallance, Exp. Physiol., 1993, 78, 303 CAS; (b) J. M. Fukuto and D. A. Wink, Met. Ions Biol. Syst., 1999, 36, 547 CAS.
- (a) S. H. Snyder and D. S. Bredt, Sci. Am., 1992, 266, 68 CrossRef CAS; (b) A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535 CrossRef CAS; (c) R. K. Pandey, J. Porphyrins Phthalocyanines, 2000, 4, 368 CrossRef CAS; (d) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897 CrossRef CAS.
- (a) P. Tavares, A. S. Pereira, J. J. G. Moura and I. Moura, J. Inorg. Biochem., 2006, 100, 2087 CrossRef CAS; (b) I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201 CrossRef CAS; (c) P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993 CrossRef CAS; (d) L. E. Goodrich, F. Paulat, V. K. K. Praneeth and N. Lehnert, Inorg. Chem., 2010, 49, 6293 CrossRef CAS.
- (a) A. Mahammed and Z. Gross, Angew. Chem., Int. Ed., 2006, 45, 6544 CrossRef CAS; (b) R. Shimanovich and J. T. Groves, Arch. Biochem. Biophys., 2001, 387, 307 CrossRef CAS; (c) S. Herold and G. Rock, Biochemistry, 2005, 44, 6223 CrossRef CAS; (d) L. M. Blomberg, M. R. A. Blomberg and P. E. M. Siegbahn, JBIC, J. Biol. Inorg. Chem., 2004, 9, 923 CrossRef CAS; (e) M. P. Jensen and D. P. Riley, Inorg. Chem., 2002, 41, 4788 CrossRef CAS; (f) J. Lee, J. A. Hunt and J. T. Groves, J. Am. Chem. Soc., 1998, 120, 7493 CrossRef CAS.
- (a) R. G. Knowles and S. Moncada, Biochem. J., 1994, 298, 249 CAS; (b) B. S. Taylor, Y. M. Kim, Q. Wang, R. A. Shapiro, T. R. Billiar and D. A. Geller, Arch. Surg., 1997, 132, 1177 CrossRef CAS.
- (a) L. K. Keefer, Curr. Top. Med. Chem., 2005, 5, 625 CrossRef CAS; (b) J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135 CrossRef CAS; (c) K. Wang, W. Zhang, M. Xian, Y. -C. Hou, X. -C. Chen, J. -P. Cheng and P. G. Wang, Curr. Med. Chem., 2000, 7, 821 CAS.
- (a) S. Maji, B. Sarkar, M. Patra, A. K. Das, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2008, 47, 3218 CrossRef CAS; (b) S. Maji, C. Chatterjee, S. M. Mobin and G. K. Lahiri, Eur. J. Inorg. Chem., 2007, 3425 CrossRef CAS; (c) P. De, B. Sarkar, S. Maji, A. K. Das, E. Bulak, S. M. Mobin, W. Kaim and G. K. Lahiri, Eur. J. Inorg. Chem., 2009, 2702 CrossRef CAS; (d) P. De, T. K. Mondal, S. M. Mobin, B. Sarkar and G. K. Lahiri, Inorg. Chim. Acta, 2010, 363, 2945 CrossRef CAS; (e) P. De, T. K. Mondal, S. M. Mobin and G. K. Lahiri, Inorg. Chim. Acta, 2011, 372, 250 CrossRef CAS; (f) G. K. Lahiri and W. Kaim, Dalton Trans., 2010, 39, 4471 RSC; (g) P. De, S. Maji, A. D. Chowdhury, S. M. Mobin, T. K. Mondal, A. Paretzki and G. K. Lahiri, Dalton Trans., 2011, 40, 12527 RSC.
- (a) N. L. Fry, M. J. Rose, D. L. Rogow, C. Nyitray, M. Kaur and P. K. Mascharak, Inorg. Chem., 2010, 49, 1487 CrossRef CAS; (b) M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093 CrossRef CAS; (c) M. J. Rose, A. K. Patra, E. A. Alcid, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2007, 46, 2328 CrossRef CAS; (d) A. K. Patra, M. J. Rose, K. A. Murphy, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2004, 43, 4487 CrossRef CAS; (e) N. L. Fry, B. J. Heilman and P. K. Mascharak, Inorg. Chem., 2011, 50, 317 CrossRef CAS; (f) M. J. Rose and P. K. Mascharak, Inorg. Chem., 2009, 48, 6904 CrossRef CAS; (g) G. M. Halpenny and P. K. Mascharak, Inorg. Chem., 2009, 48, 1490 CrossRef CAS; (h) M. J. Rose and P. K. Mascharak, Chem. Commun., 2008, 3933 RSC; (i) M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093 CrossRef CAS; (j) M. J. Rose, N. L. Fry, R. Marlow, L. Hinck and P. K. Mascharak, J. Am. Chem. Soc., 2008, 130, 8834 CrossRef CAS; (k) M. J. Rose, M. M. Olmstead and P. K. Mascharak, Polyhedron, 2007, 26, 4713 CrossRef CAS; (l) G. M. Halpenny, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2007, 46, 6601 CrossRef CAS; (m) M. J. Rose, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 2007, 129, 5342 CrossRef CAS; (n) I. Szundit, M. J. Rose, I. Sen, A. A. Eroy-Reveles, P. K. Mascharak and O. Einarsdottir, Photochem. Photobiol., 2006, 82, 1377 CrossRef; (o) A. K. Patra and P. K. Mascharak, Inorg. Chem., 2003, 42, 7363 CrossRef CAS.
- (a) C. Hauser, T. Glaser, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2000, 122, 4352 CrossRef CAS; (b) R. G. Serres, C. A. Grapperhaus, E. Bothe, E. Bill, T. Weyhermüller, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2004, 126, 5138 CrossRef CAS; (c) F. Roncaroli, M. Videla, L. D. Slep and J. A. Olabe, Coord. Chem. Rev., 2007, 251, 1903 CrossRef CAS; (d) J. A. Olabe, Dalton Trans., 2008, 3633 RSC; (e) J. A. Olabe, Adv. Inorg. Chem., 2004, 55, 61 CrossRef CAS; (f) A. Wanat, T. Schneppensieper, G. Stochel, R. van Eldik, E. Bill and K. Wieghardt, Inorg. Chem., 2002, 41, 4 CrossRef CAS; (g) J. C. Toledo, B. dos Santos Lima Neto and D. W. Franco, Coord. Chem. Rev., 2005, 249, 419 CrossRef CAS; (h) F. Di Salvo, N. Escola, D. A. Scherlis, A. Estrin, C. D. Bondia, D. Murgida, J. M. Ramallo-Lopez, F. G. Requejo, L. Shimon and F. Doctorovich, Chem.–Eur. J., 2007, 13, 8428 CrossRef CAS; (i) F. Doctorovich and F. Di Salvo, Acc. Chem. Res., 2007, 40, 985 CrossRef CAS; (j) L. L. Perissinotti, D. A. Estrin, G. Leitus and F. Doctorovich, J. Am. Chem. Soc., 2006, 128, 2512 CrossRef CAS; (k) J.-E. Jee, S. Eigler, N. Jux, A. Zahl and R. van Eldik, Inorg. Chem., 2007, 46, 3336 CrossRef CAS; (l) A. Franke, N. Hessenauer-Ilicheva, D. Meyer, G. Stochel, W. -D. Woggon and R. van Eldik, J. Am. Chem. Soc., 2006, 128, 13611 CrossRef CAS; (m) J. E. Jee, M. Wolak, D. Balbinot, N. Jux, A. Zahl and R. van Eldik, Inorg. Chem., 2006, 45, 1326 CrossRef CAS; (n) A. Franke, G. Stochel, N. Suzuki, T. Higuchi, K. Okuzono and R. van Eldik, J. Am. Chem. Soc., 2005, 127, 5360 CrossRef CAS; (o) A. Gerli, J. Reedijk, M. T. Lakin and A. L. Spek, Inorg. Chem., 1995, 34, 1836 CrossRef CAS; (p) T. E. Bitterwolf, Coord. Chem. Rev., 2006, 250, 1196 CrossRef CAS; (q) P. C. Ford and L. E. Laverman, Coord. Chem. Rev., 2005, 249, 391 CrossRef CAS; (r) K. G. Caulton, Coord. Chem. Rev., 1975, 14, 317 CrossRef CAS.
- (a) P. Singh, M. Sieger, J. Fiedler, C-Y. Su and W. Kaim, Dalton Trans., 2008, 868 RSC; (b) P. Singh, A. K. Das, B. Sarkar, M. Niemeyer, F. Roncaroli, J. A. Olabe, J. Fiedler, S. Záliš and W. Kaim, Inorg. Chem., 2008, 47, 7106 CrossRef CAS; (c) P. Singh, J. Fiedler, S. Záliš, C. Duboc, M. Niemeyer, F. Lissner, T. Schleid and W. Kaim, Inorg. Chem., 2007, 46, 9254 CrossRef CAS; (d) P. Singh, B. Sarkar, M. Sieger, M. Niemeyer, J. Fiedler, S. Záliš and W. Kaim, Inorg. Chem., 2006, 45, 4602 CrossRef CAS; (e) M. Videla, J. S. Jacinto, R. Baggio, M. T. Garland, P. Singh, W. Kaim, L. D. Slep and J. A. Olabe, Inorg. Chem., 2006, 45, 8608 CrossRef CAS; (f) A. G. De Candia, P. Singh, W. Kaim and L. D. Slep, Inorg. Chem., 2009, 48, 565 CrossRef CAS; (g) M. Sieger, B. Sarkar, S. Záliš, J. Fiedler, N. Escola, F. Doctorovich, J. A. Olabe and W. Kaim, Dalton Trans., 2004, 1797 RSC; (h) S. Frantz, B. Sarkar, M. Sieger, W. Kaim, F. Roncaroli, J. A. Olabe and S. Zálisˇ, Eur. J. Inorg. Chem., 2004, 2902 CrossRef CAS; (i) B. Mondal, H. Paul, V. G. Puranik and G. K. Lahiri, J. Chem. Soc., Dalton Trans., 2001, 481 RSC; (j) N. Chanda, D. Paul, S. Kar, S. M. Mobin, A. Datta, V. G. Puranik, K. K. Rao and G. K. Lahiri, Inorg. Chem., 2005, 44, 3499 CrossRef CAS; (k) S. Sarkar, B. Sarkar, N. Chanda, S. Kar, S. M. Mobin, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2005, 44, 6092 CrossRef CAS; (l) A. K. Das, B. Sarkar, C. Duboc, S. Strobel, J. Fiedler, S. Záliš, G. K. Lahiri and W. Kaim, Angew. Chem., Int. Ed., 2009, 48, 4242 CrossRef CAS; (m) N. Chanda, S. M. Mobin, V. G. Puranik, A. Datta, M. Niemeyer and G. K. Lahiri, Inorg. Chem., 2004, 43, 1056 CrossRef CAS.
- (a) K. Karidi, A. Garoufis, N. Hadjiliadis, M. Lutz, A. L. Spek and J. Reedijk, Inorg. Chem., 2006, 45, 10282 CrossRef CAS; (b) B. Serli, E. Zangrando, T. Gianferrara, L. Yellowlees and E. Alessio, Coord. Chem. Rev., 2003, 245, 73 CrossRef CAS; (c) E. Tfouni, M. Krieger, B. R. McGarvey and D. W. Franco, Coord. Chem. Rev., 2003, 236, 57 CrossRef CAS; (d) P. S. Ford and L. E. Laverman, Coord. Chem. Rev., 2005, 249, 391 CrossRef CAS; (e) M. Wolak and R. v. Eldik, Coord. Chem. Rev., 2002, 230, 263 CrossRef CAS; (f) S. P. Fricker, E. Slade, N. A. Powell, O. J. Vaughn, G. R. Henderson, S. A. Murrer, I. C. Megson, S. K. Bisland and F. W. Flitney, Br. J. Pharmacol., 1997, 122, 1441 CrossRef CAS; (g) S. Kuwata, S. Kura and T. Ikariya, Polyhedron, 2007, 26, 4659 CrossRef CAS; (h) M. Iwamoto and H. Hamada, Catal. Today, 1991, 10, 57 CrossRef CAS.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339 CrossRef CAS.
- A. D. Chowdhury, A. Das, K. Irshad, S. M. Mobin and G. K. Lahiri, Inorg. Chem., 2011, 50, 1775 CrossRef.
- H. Hadadzadeh, M. C. DeRosa, G. P. A. Yap, A. R. Rezvani and R. J. Crutchley, Inorg. Chem., 2002, 41, 6521 CrossRef CAS.
- S. -C. Chan, P. -K. Pat, T. -C. Lau and C. -Y. Wong, Organometallics, 2011, 30, 1311 CrossRef CAS.
- (a) F. Neese, W. Ames, G. Christian, M. Kampa, D. G. Liakos, D. A. Pantazis, M. Roemelt, P. Surawatanawong and S. Ye, Adv. Inorg. Chem., 2011, 62, 301 CrossRef; (b) J. Conradie, D. A. Quarless, H. -F. Hsu, T. C. Harrop, S. J. Lippard, S. A. Koch and A. Ghosh, J. Am. Chem. Soc., 2007, 129, 10446 CrossRef CAS; (c) A. Ghosh, Acc. Chem. Res., 2005, 38, 943 CrossRef CAS; (d) P. Ghosh, K. Stobie, E. Bill, E. Bothe, T. Weyhermu1ller, M. D. Ward, J. A. McCleverty and K. Wieghardt, Inorg. Chem., 2007, 46, 522 CrossRef CAS.
- (a) M. Kaupp, M. Bühl and V. G. Malkin, ed., Calculations of EPR g-Tensors with Density Functional Theory In Calculation of NMR and EPR Parameters, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) C. Remenyi and M. Kaupp, J. Am. Chem. Soc., 2005, 127, 11399 CrossRef CAS.
- (a) K. Ghosh, S. Kumar, R. Kumar, U. P. Singh and N. Goel, Inorg. Chem., 2010, 49, 7235 CrossRef CAS; (b) K. Ghosh, S. Kumar, R. Kumar, U. P. Singh and N. Goel, Organometallics, 2011, 30, 2498 CrossRef CAS.
- (a) A. Denicola and R. Radi, Toxicology, 2005, 208, 273 CrossRef CAS; (b) J. T. Groves, Curr. Opin. Chem. Biol., 1999, 3, 226 CrossRef CAS; (c) P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315 CrossRef CAS.
- P. R. Gardner, A. M. Gardner, L. A. Martin and A. L. Salzman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10378 CrossRef CAS.
- (a) S. Goldstein, J. Lind and G. Merenyi, Chem. Rev., 2005, 105, 2457 CrossRef CAS; (b) S. Herold and W. H. Koppenol, Coord. Chem. Rev., 2005, 249, 499 CrossRef CAS; (c) H. Gunaydin and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10036 CrossRef CAS.
- (a) L. L. Pearce, A. J. Kanai, L. A. Birder, B. R. Pitt and J. Peterson, J. Biol. Chem., 2002, 277, 13556 CrossRef CAS; (b) D. D. Thomas, M. G. Espey, M. P. Vitek, K. M. Miranda and D. A. Wink, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12691 CrossRef CAS; (c) A. G. McBride, V. Borutaite and G. C. Brown, Biochim. Biophys. Acta, 1999, 1454, 275 CAS; (d) A. G. Estevez, J. P. Crow, J. B. Sampson, C. Reiter, Y. Zhuang, G. J. Richardson, M. M. Tarpey, L. Barbeito and J. S. Beckman, Science, 1999, 286, 2498 CrossRef CAS; (e) M. Kirsch and H. de Groot, J. Biol. Chem., 2002, 277, 13379 CrossRef CAS.
- (a) M. P. Schopfer, B. Mondal, D. -H. Lee, A. A. N. Sarjeant and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 11304 CrossRef CAS; (b) G. Y. Park, S. Deepalatha, S. C. Puiu, D.-H. Lee, B. Mondal, A. A. N. Sarjeant, D. D. Rio, M. Y. M. Pau, E. I. Solomon and K. D. Karlin, JBIC, J. Biol. Inorg. Chem., 2009, 14, 1301 CrossRef CAS; (c) Y. V. Geletti, A. J. Bailey, E. A. Boring and C. L. Hill, Chem. Commun., 2001, 1484 RSC; (d) N. G. Tran, H. Kalyvas, K. M. Skodje, T. Hayashi, P. M. Loccoz, P. E. Callan, J. Shearer, L. J. Kirschenbaum and E. Kim, J. Am. Chem. Soc., 2011, 133, 1184 CrossRef CAS.
- (a) B. P. Sullivan, J. M. Calvert and T. J. Meyer, Inorg. Chem., 1980, 19, 1404 CrossRef CAS; (b) W. Kaim, S. Ernst and V. Kasack, J. Am. Chem. Soc., 1990, 112, 173 CrossRef CAS.
- G. M. Sheldrick, SHELX-97, Program for Crystal Structure Solution and Refinement, University of Göttingen: Göttingen, Germany, 1997 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1998, 37, 785 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven Jr., K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03(Revision C.02): Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- T. H. Dunning and P. J. Hay Jr., in Modern Theoretical Chemistry, ed. H. F. Schaefer, III, Pleum, New York, 1976, p 1 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- (a) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS; (b) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- (a) N. M. O'Boyle, GaussSum 2.1, 2007 Search PubMed , available at http://gausssum.sf.net; (b) N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef CAS.
- (a) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, version 3.1, Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2001 Search PubMed; (b) A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736 CrossRef CAS; (c) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS; (d) F. Weinhold, in Encyclopedia of Computational Chemistry, ed. P. V. R. Schleyer, Wiley: New York, 1998, p 1792 Search PubMed.
- D. A. Zhurko and G. A. Zhurko, ChemCraft 1.5; Plimus: San Diego, CA, 92130, http://www.chemcraftprog.com. Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details of the complexes, crystallographic material and DFT results (Table S1–S7 and Fig. S1–S9). CCDC reference numbers 838835 and 838836. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra00953f |
| This journal is © The Royal Society of Chemistry 2012 |