Kinetics of fractionation by SO2–ethanol–water (SEW) treatment: understanding the deconstruction of spruce wood chips†
Mikhail
Iakovlev
a and
Adriaan van
Heiningen
*ab
aAalto University School of Chemical Technology, Department of Forest Products Technology, POB 16300, 00076, Aalto, Finland
bUniversity of Maine, Department of Chemical and Biological Engineering, 5737 Jenness Hall, Orono, ME 04469-5737, USA. E-mail: AvanHeiningen@umche.maine.edu; Fax: 1-207-5812323; Tel: 1-207-5812278
First published on 20th February 2012
Abstract
The kinetics of SO2–ethanol–water (SEW) fractionation of spruce were studied using wood meal and chips and compared to those of SO2–water, acid sulfite (AS) and ethanol–acid sulfite treatment. The SEW lignin removal rate was found to be similar to that of AS at the same free SO2 concentration, while the lignin sulfonation rate is considerably higher for the acid sulfite systems. No relation between acidity and sulfonation rate was observed putting into question the acid-catalysed nature of this reaction. The observed SEW sulfonation and delignification patterns are consistent with Hägglund's “fast sulfonation–slow hydrolysis” consecutive scheme. The data indicate that during the initial phase hemicelluloses are removed together with lignin as lignocarbohydrate complexes, while cellulose is protected from hydrolytic attack by lignin leading to a lower hydrolysis rate. The SEW hemicellulose dissolution behaviour can be understood by the low tendency of glucomannan to “crystallise” onto cellulose. The understanding of the dissolution pattern of lignin and hemicellulose may help to interpret the enzymatic hydrolysis behaviour of SEW residual solids subjected to different degree of fractionation.
1 Introduction
The first challenge for the development of a lignocellulosic biorefinery is to fractionate biomass into its principal constituents; cellulose, hemicellulose and lignin.1,2 Spruce is a softwood, the lignocellulosic biomass which is known to be most resistant to fractionation.3 The main components of spruce are lignin (hydrophobic polyphenylpropane network, about 28%), cellulose (hydrophilic and highly crystalline linear polyglucoside, about 40%) and hemicelluloses (mostly linear short-chain polysaccharides, about 28%). Lignin and polysaccharides are bound to each other by covalent bonds including benzyl ether, benzyl ester and phenylglycosidic linkages.4 According to Lawoko et al.5 all lignin in spruce is covalently bound to carbohydrates, mostly to galactoglucomannan and arabino-4-O-methylglucuronoxylan. It was shown4,6 that lignin is bound through galactose units to the former and through arabinose, 4-O-methylglucuronic acid and xylose units to the latter. The covalent bonds between the wood components impede clean fractionation. However, this is only part of the explanation why it is difficult to deconstruct wood; the other major obstacle is the heterogeneous arrangement of the components, i.e. the ultrastructure of the fibre wall. A description of the organisation of the components in the secondary wall of softwood fibres was given by Salmén and Olsson7 and later further refined by Lawoko et al.8 In this model the cellulose microfibrils are coated by (galacto)glucomannan, while the space between the different microfibrils is filled with separate regions of lignin, (galacto)glucomannan and xylan. A heterogeneous lignin–xylan network is located midway between the microfibrils, sandwiched between the heterogeneous lignin–glucomannan network. This complex composite morphology is Nature's answer to create a tree both with strength and microbiological protection properties needed for high vertical and sustained growth. However at the same time it explains the difficulty to find a fractionation process which is able to deconstruct lignocellulosic biomass cleanly in its separate components.Fractionation of lignocellulosics leads to release of cellulosic fibres (for pulp-based products) and opens the cell wall structure by dissolution of lignin and hemicellulose between the cellulose microfibrils. The fibres are now better accessible for hydrolysis by enzymes. When the sugars in lignocellulosics are used as feedstock for fermentation, the process to open-up the cell wall structure is called pretreatment. Pretreatment has been intensively studied because it is the most important step affecting production cost of lignocellulosic ethanol.9 While access to cellulose by enzymes is the primary goal of pretreatment, clean separation of the major lignocellulosic polymers is the key objective of fractionation.1 There is however a continuum between the two processes; from steam explosion – dilute acid hydrolysis – SPORL (Na2SO3 and H2SO4 treatment plus mechanical refining10) – Lignol (ethanol–water with H2SO411) – AVAP (ethanol–water with SO212). Of all lignocellulosics, enzymatic hydrolysis of pretreated softwood biomass is most difficult.13 Despite many pretreatment studies, it is still not possible to predict the enzymatic hydrolysis behaviour of pretreated biomass based on their chemical and physical composition.14 Early work suggested that cellulose accessible surface area is one of the most important factors influencing the rate and extent of enzymatic hydrolysis of lignocellulosic substrates.15 Arantes and Saddler16 studied steam exploded (plus SO2) and ethanol treated lignocellulosic softwoods (Douglas fir and Lodgepole pine). They found that the minimum enzyme loading for efficient hydrolysis was controlled by limited accessibility of the enzymes to the cellulose chains due to the porosity/topology of the available cellulose, and not only by the external cellulose surface area. Varnai et al.17 found that lignin and/or lignin–carbohydrate complexes (LCCs) were the major component restricting the enzymatic hydrolysis of steam-pretreated (plus SO2) spruce. After chlorite delignification the hydrolysis of cellulose itself was the major limiting factor. Xylan also restricted the hydrolysis of cellulose despite its low content in the pretreated and delignified spruce, suggesting that the location of xylan also plays an important role. These findings are compatible with the earlier described ultrastructure of softwood where enzyme access (“representative” cellulase diameter of 5.1 nm16) to the spruce microfibrils (width ∼4 nm) arranged in aggregates (width ∼16 nm18) is blocked by LCCs in the lamellar space (∼10 nm19). During fractionation the polymers in the lamellar space are dissolved creating macropores between the lamellae, and intralamellar micropores.20,21 The effect of xylan reported by Varnai et al.17 is surprising since softwood microfibrils are tightly coated by glucomannan. However, Fahlen and Salmén19 found that treatment of spruce holocellulose with mannanase or xylanase dissolved both xylan and glucomannan. Knowledge of the kinetics of delignification and removal of hemicelluloses (glucomannan and xylan) both separately and as LCCs will help to understand the efficiency of enzymatic hydrolysis when applied to lignocellulosic solid residues obtained under different fractionation conditions.
A highly efficient fractionation method for lignocellulosic biomass is treatment by an ethanol–water mixture with dissolved SO2 at modest temperatures (130–160 °C).22–25 The following characteristics make the SO2–ethanol–water (SEW) process uniquely suitable for lignocellulosics fractionation. It is the only process which is able to digest different biomass species – softwoods, hardwoods and annual plants – at close delignification rates.23,26,27 Due to the very high rate of ethanol–water transport into the fibres, neither a separate impregnation stage nor small wood particles are needed, thereby minimising fractionation time and (electrical) energy use. The retention of hemicelluloses in the solid phase, as well as the cellulose degree of polymerisation as function of delignification, can be adjusted by changing process temperature and duration.22,23 About half of the dissolved hemicelluloses are hydrolysed in the liquid phase to monosaccharides, and most importantly are neither dehydrated nor oxidised.24 During recovery of ethanol and SO2 from the spent fractionation liquid the hemicelluloses are almost fully hydrolysed to monomers, thereby eliminating costly acid or enzymatic hydrolysis for this wood component.28 Cellulose is fully retained in the solid phase and can subsequently be hydrolysed to glucose. Alternatively the released cellulosic fibres may be used for conventional pulp products, either paper,29 tissue29 or dissolving23 grades. The hemicellulose monosugars may be used for production of biofuels and chemicals.30 The SEW fractionation technology is employed by American Process Inc. in a patent pending Biorefinery process termed AVAP™.12
In the present paper the kinetics of SEW fractionation are studied as a function of SO2 concentration in the fractionation liquor and are also compared to those of SO2–water, ethanol–acid sulfite and acid sulfite (AS) treatment, the latter being the second most important commercial pulping process. The kinetics are used to develop an understanding of the dissolution pattern of the three major wood components from the heterogeneous lignocellulosic structure represented by spruce wood.
2 Materials and methods
2.1 Wood meal fractionation
SO2–water, SEW, sodium–acid sulfite (AS) and ethanol sodium–acid sulfite (ethanol–AS) fractionation was performed on spruce meal (diameter 1.0 mm) to eliminate diffusion effects. The fresh cooking liquors (Table 1) were prepared by injecting gaseous sulfur dioxide into cold water, 55 v/v.% ethanol–water, aqueous sodium hydroxide and sodium hydroxide in 55 v/v.% ethanol–water solutions, respectively. Deionised water and ethanol ETAX A (96.1 v/v.%) were used. Approximately the same free SO2 concentration (12.0%) was obtained in all cases.| Liquor | Solvent | Total SO2 in fresh liquor | Bound SO2 (SO2 in HSO3− form) in fresh liquora | Free SO2 in fresh liquor | Calc. acidity in water at 135 °Cb | |||
|---|---|---|---|---|---|---|---|---|
| % | mol L−1 | % | mol L−1 | % | mol L−1 | [H+]/mmol L−1 | ||
| a The amount of bound SO2 given corresponds to the amount of sodium hydroxide and reflects the actual content of hydrosulfite anions during cooking only for the AS system. In SO2–water liquor at 135 °C the actual hydrosulfite anion concentration is about 50 mmol L−1. In SEW and ethanol–AS cooking the hydrosulfite anion concentration is unknown. b The effect of ethanol on the acidity is not considered. Acidity is given for the liquid phase. | ||||||||
| SO2–water | Water | 12.0 | 1.98 | 0 | 0 | 12.0 | 1.98 | 37 |
| SEW | 55 v/v.% ethanol–water | 12.0 | 1.82 | 0 | 0 | 12.0 | 1.82 | 36 |
| AS | Water | 13.9 | 2.33 | 1.65 | 0.28 | 12.2 | 2.05 | 5 |
| Ethanol–AS | 55 v/v.% ethanol–water | 13.9 | 2.15 | 1.82 | 0.28 | 12.1 | 1.87 | 5 |
25 o.d. g spruce meal (dry matter content 95.5%) and liquors at a liquor-to-wood ratio 4.8 L kg−1 were put into 220 mL bombs. The bombs were put into a thermostat bath at 25 °C. The impregnation at 25 °C was done for 15 h, the bombs were rotated during this period. The cooking was accomplished at 135 °C (± 1 °C) and 80 min including 8–9 min for heat-up23 and the bombs were rapidly removed from the bath and put into cold water. After cooling, the liquid phases were separated from the solid residue using a nylon washing bag. The solid residues obtained after SEW and ethanol–AS cooking were washed twice with 40% ethanol–water (at a liquid-to-solid ratio of about 4 L kg−1 and 60 °C) and once with deionised water (at a liquid-to-solid ratio of about 40 L kg−1 and 25 °C). The solid residues obtained after SO2–water and AS cooking were washed thrice with deionised water (twice at a liquid-to-solid ratio of about 4 L kg−1 and 60 °C, and once at a liquid-to-solid ratio of about 40 L kg−1 and 20 °C). Yield and kappa number31a (measure of lignin content) were determined.
Sulfur content of the solid phases was determined by combustion of 50–200 mg of the sample in oxygen in a Schöniger flask containing 0.5 mL 30% hydrogen peroxide in 25 mL water. Sample wrappers were made of Whatman 40 ashless filter paper with sulfur content 30 μg g−1 which was subtracted from the results. After absorbing the products (45 min) the content of the flask was transferred to a 100 mL measuring flask together with the washings with water. Sulfate content was analysed by ion chromatography.31b The relative error was less than 1%.
2.2 Wood chips SEW fractionation
Air dried spruce chips (dry matter content 92.9%) were screened using screens O45, //8, //6, //4 and //2 mm. The fractions from the screens //4 and //2 mm were combined and used for SEW cooking. The concentrations of SO2 in the liquors were 3.0, 6.0, 12, 18 and 27% (by weight). 25.0 o.d. (oven-dry) g of the chips and the liquor at a liquor-to-wood ratio of 6 L kg−1 were placed in 220 mL bombs. The same cooking procedure was applied for wood chips as for wood meal except that no impregnation was used, i.e. the bombs were directly put in the thermostat bath at 135 °C. The solid residues were washed twice with 50 mL (about 4 L kg−1) of the 40 v/v.% ethanol–water solution at 60 °C and finally twice with 500 mL (about 40 L kg−1) of deionised water at room temperature.22The solid residues were analysed for yield and those which have passed the defibration point, i.e. pulps, were analysed also for kappa number and intrinsic viscosity in cupriethylenediamine (CED) solution.31c The latter was measured for the air-dried unbleached pulps not later than after two weeks of storage at room temperature. Solid residues with kappa number higher than 35 were subjected to chlorite delignification.32 Cellulose viscosity-average DP was calculated according to the following formula:33
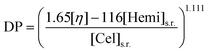 | (1) |
It was noticed that the intrinsic viscosity substantially decreases during storage of air-dried non-bleached solid residues at room temperature (one year storage leads to about 30–40% decrease in intrinsic viscosity). In the air-dried solid residues the amount of liquid contained in the solids is much lower than that during fractionation (around 0.05 g water/g solid residue compared to about 1.4 g water/g solid residue, the latter being the fibre saturation point of the solid residues29). That means that the concentration of hydroxonium cations generated by lignosulfonic acids substantially increases upon drying, and the high acidity promotes cellulose hydrolysis. Therefore the air-dried unbleached solid residues should not be stored for a long time prior to viscosity measurement.
Lignin, carbohydrate and sulfur content of the wood and solid residues was measured after acetone extraction.31d
The lignin content of the wood and the solid residues prepared using 12% SO2 liquor was measured according to refs. 34 and 35 while the lignin content of the defibrated solid residues (pulps) prepared using 3.0, 6.0, 18 and 27% SO2 liquors as well as the solid residues prepared from wood meal was calculated from the kappa (κ) number values using the relationship: [Lig] = 0.165κ + 0.63.35
The carbohydrate content of the wood and solid residues was determined using two techniques: acid methanolysis with GC-FID detection based on Sundberg et al.36 and double-stage sulfuric acid hydrolysis with HPAEC-PAD detection according to refs. 24 and 34.
SO2 concentration in the liquid phase was measured according to the SCAN standard31e (note that the sample should be immediately contacted with alkali to avoid SO2 losses).
The sulfonation degree of lignin (S/C9 molar ratio) was calculated based on a molecular weight of softwood lignin monomer of 190 g/mol.37a
3 Results and discussion
3.1 Wood meal fractionation: the chemistry of SEW vs. other SO2-based processes
To better understand the chemistry of SO2–ethanol–water (SEW) fractionation it was compared to SO2–water, acid sulfite (AS) and ethanol acid sulfite (ethanol–AS) cooking, all performed at 12% free SO2, 135 °C and 80 min. By using wood meal and applying 15 h impregnation at room temperature the influence of diffusion on the rate of the processes was minimised. Composition of the solid residues is given in Table 2.| Process | Colour | Yield (%) | Kappa | Lignin % on wood | LFY % on wood | Sulfur in solid residue | Sulfur in liquid phase | Total accumulated sulfur | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| % on wood | S/C9 | % on wood | S/C9 | % on wood | S/C9 | ||||||
| a L/W ratio 6 L kg−1. | |||||||||||
| SO2–water | Brownish | 56.9 | 122 | 11.8 | 45.1 | 0.323 | 0.162 | 1.32 | 0.493 | 1.64 | 0.352 |
| SEW | Bright | 53.0 | 34.3 | 3.33 | 49.7 | 0.0685 | 0.122 | 0.813 | 0.198 | 0.882 | 0.189 |
| AS | Extremely bright | 50.9 | 21.5 | 2.13 | 48.8 | 0.125 | 0.349 | 2.57 | 0.598 | 2.70 | 0.579 |
| Ethanol–AS | Very bright | 85.6 | 98.0 | 14.4 | 71.2 | 0.812 | 0.335 | 1.34 | 0.597 | 2.15 | 0.461 |
| SEW, chipsa | Bright | 51.2–51.8 | 29.4–33.5 | 2.81–3.30 | 48.3–48.7 | 0.0476 | 0.086 | 0.94–1.08 | 0.23–0.26 | 0.99–1.13 | 0.21–0.24 |
It can be seen that the solid residue and liquid phase obtained in the present SEW fractionation of spruce meal has nearly the same composition and sulfur content as that obtained in SEW cooking of the chips without separate impregnation. It again proves that impregnation is not necessary in SEW fractionation and that diffusion of SO2 is not a rate-limiting factor for this process.
The LFY is controlled by the degree of hydrolysis and dissolution of the carbohydrates. Thus LFY can be considered as a measure of the effective acidity of the fractionation system at temperature. Highly acidic SO2–water cooking intensively removes carbohydrates (LFY 45.1%), although the delignification rate is relatively low (residual lignin of 11.8% on wood). On the other hand the LFY of low acidic ethanol–AS pulping (both base and ethanol are present) is the same as that in the original wood. By comparing LFY of AS (48.8%) and SEW (49.7%) pulps one can conclude that the effective acidity of these systems is similar. However, the calculated acidity at the cooking temperature of SEW is about 7 times higher than that of AS when the effect of ethanol on acidity is neglected (see Table 1). Thus it follows that the presence of ethanol at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) ratio in water significantly decreases the effective acidity. This is in agreement with the finding that addition of ethanol to water suppresses dissociation of sulfurous acid due to decreased stabilisation of the formed ions in less polar solvents.38
:1 (w/w) ratio in water significantly decreases the effective acidity. This is in agreement with the finding that addition of ethanol to water suppresses dissociation of sulfurous acid due to decreased stabilisation of the formed ions in less polar solvents.38
It should be added that the considerably lower sulfonation degree of SEW lignin at which its dissolution becomes possible may be related to the significantly higher solubility of the lignosulfonic acids in ethanol–water compared to that of lignosulfonates in the aqueous AS liquor.42,43
Absence of a straightforward correlation between S/C9 ratio (corresponding to sulfonation/sulfitolysis) and delignification rate suggests that delignification is determined by another chemical reaction, probably hydrolysis. Assuming hydrolysis to be the rate-determining step as in Hägglund's consecutive sulfonation–hydrolysis scheme,45 it would be easy to explain the difference in lignin dissolution rates based on the differences in effective acidities of the liquors. The effective acidity of the systems is decreasing in the order SO2–water > AS ≈ SEW > ethanol–AS. The lignin dissolution rate follows the same order with the exception of the SO2–water system which is considerably affected by condensation.
The above comparison of the four fractionation systems shows that there is a balance between lignin sulfonation, condensation and hydrolysis reactions. Thus there exists an optimum acidity and sulfonating capacity for the fractionation systems at which hydrolysis is maximised and condensation is minimised. This optimum condition was found empirically for AS and SO2–water cooking by varying bound and free SO2 amounts (so-called Kaufmann diagram).37c The same optimum exists for the SEW system and was empirically found to occur at a 1:1 ethanol–water ratio;46 with a higher ratio leading to a too low acidity for efficient hydrolysis, while at a lower ratio the acidity is too high leading to significant condensation.
3.2 Chips cooking
The applied cooking liquors contain high weight percentages of SO2 (3.0–27%). Due to the relatively high pKa value of sulfurous acid at cooking temperature (3.2 in water at 135 °C;37d higher in ethanol being a weaker proton acceptor compared to water), the dissociated amount of SO2 can be neglected, and the concentration of SO2 in the fresh liquor at cooking temperature can directly be calculated from the charged amount of SO2. As cooking proceeds SO2 is consumed in the reactions with lignin and in side reactions. However, the highest measured amount of bound sulfur corresponds to only 1.1% on wood with the rest recoverable as SO2.24 Therefore the concentration of SO2 in the liquid phase is assumed to be constant during cooking and equal to that in the fresh liquor.
The acidity during cooking may be estimated from the amounts of sulfur dioxide and formed strong lignosulfonic acid groups (effect of ethanol is neglected). The initial concentration of SO2 in the cooking liquor is 0.43 (3.0%), 0.87 (6.0%) and 1.79 (12%) mol L−1. At 135 °C according to the pKa1 of sulfurous acid in water, the hydroxonium cation concentrations for the different SO2 concentrations are 0.02, 0.02 and 0.03 mol L−1, respectively. The amounts of lignosulfonic acids formed during cooking at these SO2 concentrations correspond to about 0.03, 0.03 and 0.05 mol L−1, respectively.24 These numbers suggest that the acidity of the liquid phase does not change significantly during SEW fractionation. This is indirectly confirmed by the pH of the liquid phases measured at room temperature (using conventional glass electrode, Table S1, ESI†) which shows changes of only 0.1–0.2 pH units during the entire fractionation process.
It is known that the acidity in the fibre-bound liquid is different from that of the free liquid outside the fibres47 when the fibre wall contains a significant amount of ionisable groups (i.e. sulfonic acids). The importance of this so-called Donnan equilibrium was estimated as follows. Assuming full ionisation of lignosulfonic acid groups attached to the solid phase, and a density of the fibre-bound liquid of 1.0 g mL−1,37e the acidity of the fibre-bound liquid would be about 0.06 M at the beginning of cooking and decrease constantly during fractionation to about 0.02 M. The acidity of the free liquid outside the fibres is calculated to be 0.02–0.05 M. Therefore the acidities of the two liquid phases, neglecting the effect of ethanol, are very close. The presence of ethanol reduces the ionisation of all acids and thus would further decrease the difference in acidity between the fibre-bound and free liquid. Thus it may be concluded that Donnan equilibrium is not important in SEW fractionation.
The concentration of hydrosulfite anions is difficult to estimate. Their concentration in the SEW fractionation liquid, when disregarding the presence of ethanol, is about 10–40 mmol L−1 at 135 °C, which is an order of magnitude lower than that in acid sulfite liquor (300 mmol L−1).48a However, since our wood meal experiments showed that the acidity of SEW liquor is similar to that of AS, this would suggest that the hydrosulfite anion concentration in SEW liquor is two orders of magnitude lower. This is an important advantage of SEW fractionation compared to AS cooking where hydrosulfite anions are responsible for the unwanted side reactions including sugar oxidation.48b It also implies that sulfur dioxide solvates are the only possible sulfonating agents in SEW fractionation.
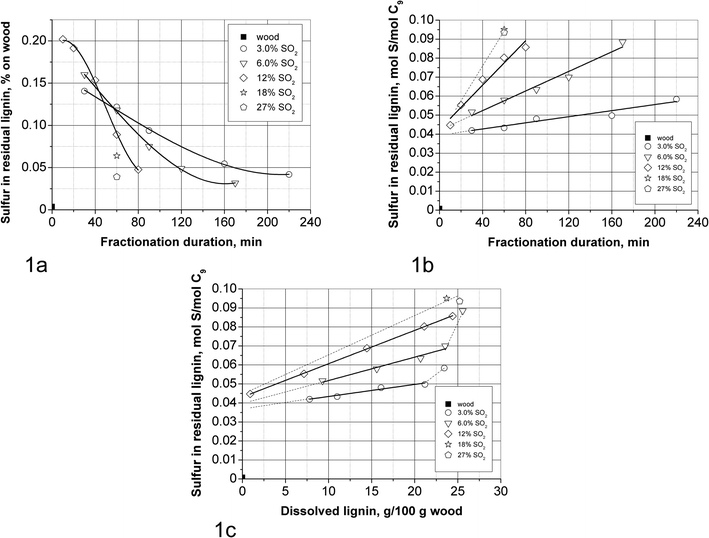 | ||
| Fig. 1 Sulfur content in the residual lignin: (a) sulfur content based on wood vs. fractionation duration; (b) S/C9 ratio vs. fractionation duration; (c) S/C9 ratio vs. amount of dissolved lignin. | ||
Fig. 1a and b show the sulfur content of the solid residues based on original wood and as S/C9 ratio vs. cooking duration, respectively. The solid phase reaches the highest sulfur content on original wood basis (about 0.2% on wood or 0.045 S/C9 for the 12% SO2 cooking) after only 10 min of cooking, i.e. at the end of the heat-up period. At this point about 97% of lignin is still retained in the solid residue. As cooking proceeds the sulfur content of the solid residues based on wood decreases due to lignin dissolution. The development over time of the sulfur content of the solid residues based on original wood shown in Fig. 1a can be compared to that in AS cooking.37f The first stage is governed by sulfonation of lignin without its dissolution (i.e. during the heat-up period in SEW and taking a few hours during impregnation of the AS process) and the second stage being lignin dissolution resulting in a rapid decrease in the sulfur content of the solid residue (durations are similar for SEW and AS processes).
However if the sulfur content is based on the residual lignin content (see Fig. 1b) then the degree of lignin sulfonation, expressed as S/C9 ratio, gradually increases by a factor 1.5–2 over the entire cook. It is noted that the S/C9 values (0.040–0.095) are considerably lower than the value of 0.3 required for lignin dissolution in AS cooking.37b Otherwise the relative development of the sulfur content over time in SEW cooking resembles that in AS cooking37f where the S/C9 also increases until the very last stages of the cook. Finally it can be seen in Fig. 1b that the rate of increase of S/C9 increases roughly linearly with SO2 concentration from 3.0 to 12%: 8.00 × 10−5 (at 3.0% SO2), 25.8 × 10−5 (at 6.0% SO2) and 58.4 × 10−5 (at 12% SO2) mol S/(mol C9 × min). No clear increase in the rate is observed between 18 and 27% SO2.
The dissolved SEW lignin has a higher S/C9 content (0.16–0.26 S/C924) which means that higher sulfonated lignin is removed preferentially from the solid phase. Nevertheless, the degree of sulfonation of dissolved lignin is also substantially lower than that in AS pulping (S/C9 ratio of 0.5–0.737b), especially at lower SO2 concentrations. A similar difference in S/C9 ratio of the dissolved SEW and AS lignin was also observed in wood meal cooking (Table 2).
The lower sulfonation degree of SEW lignin is explained by the near absence of hydrosulfite anions being more reactive nucleophiles compared to the sulfur dioxide solvates.
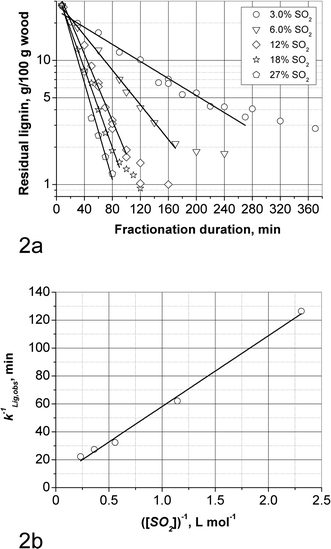 | ||
| Fig. 2 SEW delignification: (a) Decimal logarithm of residual lignin based on wood vs. fractionation duration; (b) The inverse of delignification rate constant, kLig.obs, vs. the inverse of SO2 concentration. | ||
| [SO2] in original liquor (%) | Delignification | Mannan removal | Xylan removal | Hemicelluloses removal | Cellulose hydrolysis |
|---|---|---|---|---|---|
| 103kLig,obs | 103kMan | 103kXyl | 103kHemi | 106kCel | |
| a Hemicelluloses content of the solid residues is measured by acid hydrolysis HPAEC-PAD and acid methanolysis GC-FID.24 b Hemicelluloses content is calculated by subtracting cellulose (assumed to be totally retained in the solid phase) and lignin content from the yield. c n.m. = not measured. | |||||
| 3.0 | 7.91 | 2.56 | 2.80 | 2.97a | 1.14 |
| 6.0 | 16.1 | 4.15 | 3.56 | 3.78a | 1.36 |
| 12 | 30.9 | 8.46 | 6.91 | 8.95a | 1.45 |
| 18 | 36.5 | n.m. | n.m. | 10.0b | 1.72 |
| 27 | 44.9 | n.m. | n.m. | 14.6b | n.m.c |
Therefore, for the concentration range of 3.0–12% SO2 and 135 °C the following equation is valid:
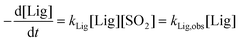 | (2) |
It can be seen in Fig. 2a that the end of the bulk delignification phase (and thus the beginning of residual delignification) occurs at higher lignin content when the SO2 concentration decreases. This may be explained by more lignin condensation at the same amount of delignification when the SO2 concentration is lower. This is supported by the observation that both the solid and liquid phases at 3.0% SO2 have a considerably more intense brown colour than those produced at higher SO2 concentrations. Also it can be seen that at 3.0% SO2 it is not possible to reach a low lignin content of about 2 g/100 g original wood. The increase of the importance of condensation at lower SO2 concentrations is understandable based on the knowledge that condensation increases with increasing acidity and decreasing sulfonation, while delignification increases with increasing SO2 concentration. Since the acidities are rather similar during the bulk delignification phase at the different SO2 concentrations, it follows that the importance of condensation increases at lower SO2 concentrations. Condensation may be the reason for the fact that at 3.0% SO2 the S/C9 ratio of the dissolved lignin is the same at 60 and 220 min of fractionation, 0.16–0.17, although 12% (on wood) of lignin is removed in between.24
According to Richards and van Heiningen,49 the kinetics of AS delignification can be described by the following equation:
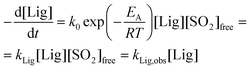 | (3) |
Therefore, at 135 °C for AS delignification kLig is equal to (23 ± 4) × 10−3 L mol−1 min−1. This value is comparable to the rate constant of SEW delignification, kLig, of (18 ± 1) × 10−3 L mol−1 min−1, and thus provides further confirmation that SO2 is the sulfonating species. It also is indirect evidence that the “bulk” delignification rate constants are not affected by condensation.
Both sulfonation and delignification rates increase considerably slower at the highest SO2 concentrations (12–27%). Therefore a physicochemical explanation for a heterogeneous reaction system was considered in which the reactant (SO2) first adsorbs on the active lignin site (α-carbon) before sulfonation takes place. The overall rate of such a process would be dependent on the amount of the available reaction sites and could be written following Langmuir–Hinschelwood kinetics as:
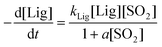 | (4) |
By plotting the inverse of the observed delignification rate constant, kLig,obs, vs. the inverse of SO2 concentration (Fig. 2b) the value for a of 0.145 L mol−1 was obtained. Therefore the term a[SO2] in the denominator equals to 0.06, 0.26 and 0.62 for SO2 concentrations of 3.0, 12 and 27%, respectively. This explains that at concentrations lower than 12% the kinetics are apparently first order in SO2.
It can be seen in Fig. 3a that the intrinsic viscosity is lower at a particular kappa number (measure of lignin content of pulp) at lower SO2 concentrations. Thus the selectivity of delignification may be improved substantially by increasing the SO2 concentration. This behaviour is similar to the selectivity response of AS delignification to the free SO2 concentration.48c
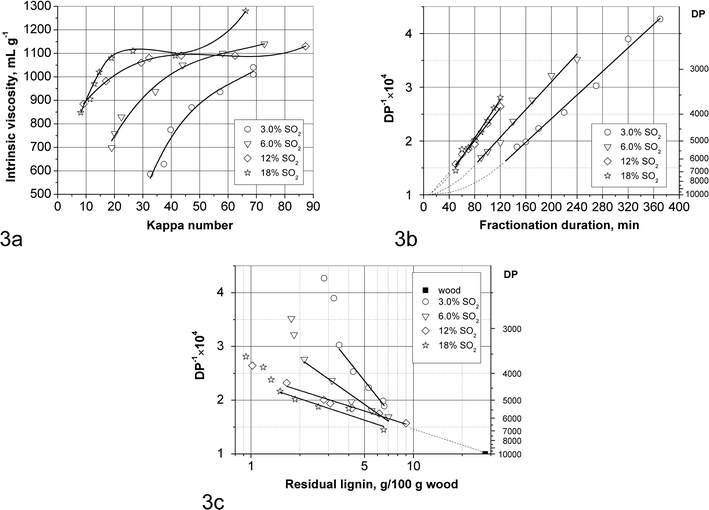 | ||
| Fig. 3 Cellulose hydrolysis in SEW fractionation: (a) solid residue intrinsic viscosity vs. kappa number; (b) the inverse of cellulose DP vs. fractionation duration; (c) the inverse of cellulose DP vs. decimal logarithm of residual lignin. | ||
The viscosity-average cellulose DP, being close to the weight-average DP, was calculated from the intrinsic viscosity using the empirical eqn (1) which also accounts for the cellulose and hemicelluloses content of the solid residues. The plot of the reverse DP vs. fractionation duration is shown in Fig. 3b. At a particular initial SO2 concentration the points lie on a straight line which proves indirectly that the acidity of the solid phase does not change significantly after the fibre liberation point as discussed in the previous section and as supported by the pH values measured for the liquid phases (Table S1, ESI†). Therefore the rate of cleavage of the cellulose chains should be proportional to the hydroxonium cation concentration as described by the following equation, Emsley and Stevens:52
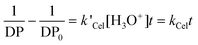 | (5) |
Also, since the slopes of these lines (i.e. kCel, Table 3) increase only 50% when increasing the SO2 concentration 6 times, it means that the SO2 concentration does not directly affect the rate.
The straight lines for 12 and 18% SO2 in the original liquor are almost identical and pass through the point (t = 9 min, DP = 10000) corresponding to the weight-average DP of spruce cellulose37g at the equivalent heat-up time. At lower SO2 concentrations on the other hand a certain delay period is seen prior to steady-state cellulose hydrolysis. To explain this phenomenon, the inverse of the cellulose DP is plotted in Fig. 3cvs. the decimal logarithm of the residual lignin. The linear fits correspond to the bulk delignification phase data and the slope is equal to 2.303kCel/kLig,obs. The increase in the slope at lower SO2 concentrations corresponds to increased cellulose hydrolysis relative to delignification, i.e. diminished selectivity. It can be seen that the straight lines for the SO2 concentrations of 3.0, 6.0 and 12% cross at a residual lignin content of 7–8% on wood. Since the original wood contains 28% lignin, this may be interpreted that during the initial phase of delignification the lignin protects cellulose from hydrolytic attack, and the cellulose DP decreases at the same rate at all SO2 concentrations until 7–8% on wood of residual lignin is left. Interestingly this lignin content corresponds to the fibre liberation point. Considering the much larger size of cellulytic enzymes (∼5 nm) compared to hydroxonium ions, this implies that for efficient enzymatic hydrolysis lignin has to be removed at least to this level corresponding to the fibre liberation point.
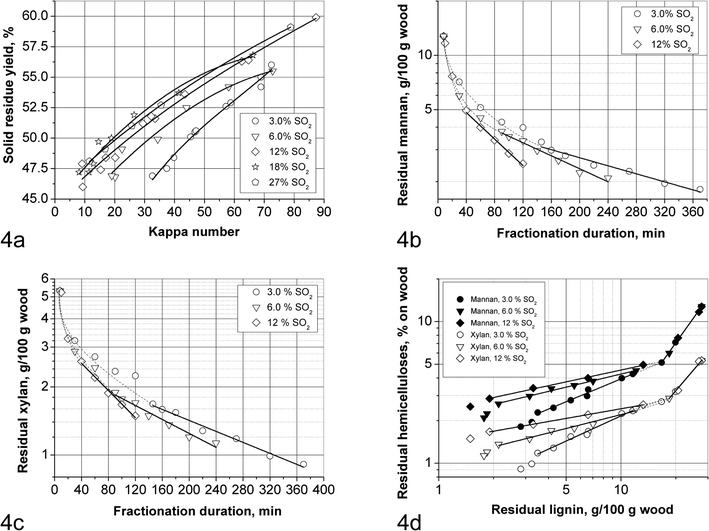 | ||
| Fig. 4 Hemicelluloses removal in SEW fractionation: (a) solid residue yield vs. kappa number; (b) decimal logarithm of residual mannan vs. fractionation duration; (c) decimal logarithm of residual xylan vs. fractionation duration; (d) decimal logarithm of residual hemicelluloses content vs. decimal logarithm of residual lignin. | ||
The decimal logarithms of mannan and xylan content of the solid residues are plotted vs. cooking duration in Fig. 4b and c, respectively. It can be observed that dissolution of the polysaccharides proceeds in two phases. In the first (initial) phase more than half of mannan and xylan is dissolved relatively quickly, while in the second phase their dissolution follows linear trends which includes delignification beyond the fibre liberation point. The latter is occurring at a lignin content of 7–9% on wood but at different mannan and xylan content for different SO2 concentrations. Furthermore, the lower the SO2 concentration in the original liquor, the longer is the first phase and more hemicelluloses are removed in this phase.
The linear trend of the second phase can be interpreted in terms of the following equation48e with the slopes being proportional to the hemicellulose dissolution rate constants (kHemi, given in Table 3):
 | (6) |
Although the rate constants are substantially lower at lower SO2 concentrations, still at lower SO2 concentrations more hemicelluloses are dissolved when reaching the fibre liberation point. So the overall effect of SO2 concentration on the hemicellulose dissolution is smaller than on delignification, and explains the appreciable difference in yield selectivity at different SO2 concentrations as shown in Fig. 4a. This is explained by the same facts as given earlier for the effect of SO2 on cellulose degradation: the acidity is similar at different SO2 concentrations, while delignification increases nearly linearly with SO2 concentration.
Further insight in the fractionation process is obtained when the hemicellulose retention is plotted vs. residual lignin content in Fig. 4d. In this case also two phases are observed but now both follow a linear behaviour with the slope being equal to the ratio of the rate constants kHemi/kLig,obs. During the initial phase (residual lignin 27.7 to 16% on wood) the relative hemicellulose dissolution rate is obviously not dependent on the SO2 concentration. This may be interpreted that during this phase hemicellulose removal and delignification are linked. Since delignification is first order in lignin from the very beginning of fractionation up to the end of bulk delignification this suggests that delignification governs the first phase of hemicellulose dissolution whereby half or more of the mannan and xylan are dissolved. In effect the solubilised lignin carries the hemicellulose with it in the form of lignin-carbohydrate complexes. On the other hand, during the second phase below 16% residual lignin the hemicellulose dissolution proceeds at lower rates relative to delignification at higher SO2 concentrations.
Mannan is removed somewhat faster than xylan in the initial phase as well as in the second phase at 6.0 and 12% SO2 (see Table 3). It can be noted also that the ratio kMan/kXyl for the second phase increases with increasing SO2 concentration. The observed behaviour is contrary to the about 1.5 times higher acid hydrolysis rate found53 for xylose glycosides compared to that for mannose glycosides. It also contradicts the higher relative retention of glucomannan in acid sulfite paper-grade pulps compared to 4-O-methylglucuronoxylan.37h,48f However, the same behaviour was observed for spruce AS dissolving pulp.48g This can be explained by the fact that the reactivity of the wood polymers does not directly correspond to that of the corresponding methylglycosides in a homogeneous solution. It is known that dissolved xylan is considerably stabilised by side 4-O-methylglucuronic acid units against acid hydrolysis both in AS and SEW cooking. On the other hand, the high wood glucomannan stability in AS cooking can be attributed to “crystallisation” onto cellulose. The latter effect is dependent on the conditions at the beginning of the treatment, and the highest retention is observed when deacetylation and removal of the galactose side units is accomplished at mild conditions without major hydrolysis of the glycoside bonds of the glucomannan backbone. On the other hand, it is said that crystallisation of xylan is hindered54 by the presence of uronic acid side units, and for softwoods by a longer diffusion path since glucomannan is mostly associated with cellulose, while xylan is more associated with lignin.7
This is supported by the following observations: 1. very high glucomannan yields in two-stage sulfite cooking with the first stage being less acidic; 2. isolated galactoglucomannan is hydrolysed faster than 4-O-methylglucuronoxylan.55
Thus it is possible that the conditions of SEW cooking characterised by the absence of impregnation at low temperature lead to a lower molecular weight of glucomannan which has a lower tendency to crystallise onto cellulose. However, at lower SO2 concentrations residual mannan is more slowly degraded possibly also due to protection by lignin in the same way as cellulose is protected from acid hydrolysis, and thus can crystallise more efficiently. The implication of the above described hemicellulose removal is that enzymatic hydrolysis of the residual solids at the same delignification below the fibre liberation point (7–9% on wood) may be improved at higher SO2 concentrations despite the higher hemicellulose content because less crystallisation of glucomannan onto cellulose has taken place.
Conclusions
The SO2–ethanol–water (SEW) fractionation experiments performed on spruce wood meal and chips revealed that:
• Diffusion of SO2 into the wood is not a rate-limiting step.
• The effective acidities of the SEW and AS cooking systems are similar, and the former is only slightly dependent on SO2 concentration and fractionation duration.
• The Donnan effect is of minor importance in SEW cooking.
• The effective acidity of the ethanol–AS cooking system is very low leading to insignificant carbohydrate dissolution.
SEW delignification is characterised by the following:
• The sulfur accumulation rate is considerably higher in the base-containing (AS and ethanol–AS) cooking systems compared to that of base-free (SO2–water and SEW) cooking due to the higher concentration (activity) of hydrosulfite anions in the former.
• There is no correlation between effective acidity and sulfonation rate at the same free SO2 concentration. This suggests that the mechanism is not acid-catalysed substitution, but perhaps nucleophilic addition of SO2 to quinone methide.
• A direct correlation between lignin sulfonation and dissolution is absent. This fact favours the Hägglund's consecutive sulfonation–hydrolysis scheme with the latter reaction being rate-determining.
• Lower lignin condensation is observed at higher SO2 concentrations and in the presence of a base. However, condensation does not seem to affect the rate of SEW bulk delignification as it is similar to AS cooking at the same free SO2 concentration.
• The rates of both delignification and residual lignin sulfonation increase linearly from 3.0 to 12% SO2, while at 18–27% SO2 this increase slows down considerably possibly due to SO2 adsorption saturation.
Carbohydrate hydrolysis/removal in SEW fractionation is characterised by the following:
• Cellulose hydrolysis and hemicellulose dissolution occur in two phases, initial and bulk. Cellulose hydrolysis is slower in the initial phase (down to 7–8% residual lignin) than in the bulk phase, while the opposite is true for the hemicellulose dissolution.
• During the initial stage both hemicellulose dissolution and cellulose hydrolysis are determined by lignin dissolution and independent of the fractionation conditions. It is suggested that lignin–carbohydrate bonds are not yet substantially cleaved, and about half of the original glucomannan and xylan are removed together with lignin. In addition, lignin protects cellulose from hydrolytic attack.
• The kinetics for the bulk phases of cellulose hydrolysis and hemicellulose dissolution are zero order in the number of glycosidic bonds and first order in residual hemicelluloses, respectively.
• The rates of bulk cellulose hydrolysis are similar at 3.0–18% SO2 as the effective acidity is not much affected when the SO2 concentration is changed over this range.
• Glucomannan is removed faster than xylan in SEW fractionation which is explained by limited stabilisation of the former compared to that seen in AS cooking. The better glucomannan dissolution is of interest when dissolved hemicelluloses are an important product as part of the fractionated sugars. At the same time the lack of stabilisation of glucomannan may improve enzymatic hydrolysis of the residual solids at the same delignification below the fibre liberation point (7–9% on wood).
• SEW delignification selectivity may be substantially improved by increasing the SO2 concentration which is similar to the AS delignification selectivity response when the free SO2 is increased.
Acknowledgements
We thank Dr. Raimo Alén for providing Schöniger combustion apparatus. The help of Lidia Testova and the discussions with Dr. Herbert Sixta and Niko Aarne are deeply acknowledged. Financial support by TEKES through the FiDiPro program is greatly appreciated.References
- J. J. Bozell, Bioresources, 2010, 5, 1326–1327 CAS.
- H. Reith, H. de Uil, R. van der Linden, W. Huijgen and J. Wildschut, NPT Procestechnologie, 2011, 18(1), 26–28 CAS.
- J. Y. Zhu, X. J. Pan, G. S. Wang and R. Gleisner, Bioresour. Technol., 2009, 100, 2411–2418 CrossRef CAS.
- D. Fengel and G. Wegener, Wood—Chemistry, Ultrastructure, Reactions, Walter de Gruyter, Berlin, 1989 Search PubMed.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Holzforschung, 2006, 60(2), 156–161 CrossRef CAS.
- O. Eriksson and B. O. Lindgren, Svensk Papperstidning, 1977, 80(2), 59–63 CAS.
- L. Salmén and A.-M. Olsson, J. Pulp Pap. Sci., 1998, 24(3), 99–103 Search PubMed.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Biomacromolecules, 2005, 6, 3467–3473 CrossRef CAS.
- R. T. Elander, B. E. Dale, M. Holtzapple, M. R. Ladisch, Y. Y. Lee, C. Mitchinson, J. N. Saddler and C. E. Wyman, Cellulose, 2009, 16, 649–659 CrossRef CAS.
- J. Y. Zhu and X. J. Pan, Bioresour. Technol., 2010, 101, 4992–5002 CrossRef CAS.
- W. E. Mabee, D. J. Gregg, C. Arato, A. Berlin, R. Bura, N. Gilkes, O. Mirochnik, X. Pan, E. K. Pye and J. N. Saddler, Appl. Biochem. Biotechnol., 2006, 129, 55–70 CrossRef.
- T. Retsina and V. Pylkkänen, Paper 360°, 2007,(February issue), 18–19 Search PubMed.
- M. Galbe and G. Zacchi, Adv. Biochem. Eng./Biotechnol., 2007, 108, 41–65 CrossRef CAS.
- R. Kumar and C. E. Wyman, Woodhead Publishing Series in Energy, 2010, 3, 73–121 CAS.
- D. N. Thompson, H. C. Chen and H. E. Grethlein, Bioresour. Technol., 1992, 39, 155–163 CrossRef CAS.
- V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2011, 4(3), 1–16 Search PubMed.
- A. Varnai, M. Siika-aho and L. Viikari, Enzyme Microb. Technol., 2010, 46, 185–193 CrossRef CAS.
- J. Fahlen and L. Salmen, Biomacromolecules, 2005, 6, 433–438 CrossRef CAS.
- J. Fahlen and L. Salmen, Holzforschung, 2005, 59, 589–597 CrossRef CAS.
- J. E. Stone and A. M. Scallan, Tappi J., 1967, 50(10), 496–501 CAS.
- J. E. Stone and A. M. Scallan, Cellul. Chem. Technol., 1968, 2(3), 343–358 CAS.
- M. Iakovlev, T. Pääkkönen and A. van Heiningen, Holzforschung, 2009, 63, 779–784 CrossRef CAS.
- M. Iakovlev, H. Sixta and A. van Heiningen, J. Wood Chem. Technol., 2011, 31, 250–266 CrossRef CAS.
- M. Iakovlev and A. van Heiningen, ChemSusChem, 2012 DOI:10.1002/cssc.201100600.
- M. Iakovlev, Doctoral thesis, Aalto University, Finland, 2011.
- S. F. Primakov, Nauchn. Tr. Vses. Nauchn.-Issled. Inst. Tsellyulozn.-Bumazhn. Prom., 1961, 47, 69–75 Search PubMed.
- M. Yamamoto, M. Iakovlev and A. van Heiningen, Holzforschung, 2011, 65, 559–565 CrossRef CAS.
- E. Sklavounos, M. Iakovlev, M. Yamamoto, A.-L. Teräsvuori, G. Jurgens, T. B. Granström and A. van Heiningen, Holzforschung, 2011, 65, 551–558 CrossRef CAS.
- M. Iakovlev, E. Hiltunen and A. van Heiningen, Nord. Pulp Pap. Res. J., 2010, 25, 428–433 CAS.
- M. Rakkolainen, M. Iakovlev, A.-L. Teräsvuori, E. Sklavounos, G. Jurgens, T. B. Granström and A. van Heiningen, Cellul. Chem. Technol., 2010, 44, 139–145 CAS.
- (a) Scandinavian Standards: SCAN-C 1:00, Chemical pulp: Kappa number, 2000; (b) SCAN-CM 57:99: Pulp: Total sulphur content, 1999; (c) SCAN-CM 15:99, Viscosity in cupriethylenediamine solution, 1999; (d) SCAN-CM 49:03, Content of acetone-soluble matter, 2003; (e) SCAN-N 36:98, Sulphite, sulphate and thiosulphate ion concentrations, 1998.
- Tappi standard, 1966. T 230. Om-66, Viscosity of pulp (capillary viscometer method).
- D. da Silva Perez and A. R. P. van Heiningen, Seventh European Workshop on Lignocellulosics and Pulp (EWLP, proceedings), 2002, 393–396 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass. Laboratory Analytical Procedure, National Renewable Energy Laboratory, Golden, CO, 2008 Search PubMed.
- M. Iakovlev and A. van Heiningen, J. Wood Chem. Technol., 2011, 31, 233–249 CrossRef CAS.
- A. Sundberg, K. Sundberg, C. Lilland and B. Holmbom, Nord. Pulp Pap. Res. J., 1996, 11, 216–219 CAS.
- S. A. Rydholm, Pulping Processes, John Wiley & Sons Inc., London, 1965: (a) p. 186, (b) p. 490, (c) p. 467, (d) p. 456, (e) p. 477, (f) p. 489, (g) p. 108, (h) p. 540 Search PubMed.
- B. L. Wedzicha and S. J. Goddard, Food Chem., 1991, 40, 119–136 CrossRef CAS.
- L. Ivnäs and B. Lindberg, Acta Chem. Scand., 1961, 15, 1081–1086 CrossRef.
- S. F. Primakov, Nauchn. Tr. Vses. Nauchn.-Issled. Inst. Tsellyulozn.-Bumazhn. Prom., 1961, 46, 83–118 Search PubMed.
- R. K. Boyarskaya and M. N. Tsypkina, Khim. Drevesiny, 1970, 5, 59–66 CAS.
- S. F. Primakov, I. M. Tsarenko and V. P. Zaplatin, Koksnes Kimija, 1979, 6, 39–42 Search PubMed.
- S. F. Primakov and V. A. Barbash, Bumazhnaya Promyshlennost, 1989, 1, 6–7 Search PubMed.
- V. Pylkkänen, Master thesis, Lappeenranta University of Technology, Finland, 1992.
- E. Hägglund, Chemistry of Wood, Academic Press Inc., New York, 1951 Search PubMed.
- M. G. Eliashberg, A. I. Parfenova and S. F. Primakov, Trudy Leningrad. Lesotekh. Akad. im. S. M. Kirova, 1960, 91, 235–245 Search PubMed.
- G. Fiehn, Zellstoff Papier (Leipzig), 1964, 13(5), 136–139 CAS.
- Handbook of Pulp, ed. H. Sixta, Wiley-VCH, Weinheim, 2006, vol. 1, pp. 109–509: (a) p. 396, (b) p. 423, (c) p. 460, (d) p. 461, (e) p. 408, (f) p. 110, (g) p. 449 Search PubMed.
- T. Richards and A. van Heiningen, J. Pulp Pap. Sci., 2004, 30(10), 263–268 CAS.
- B. D. Favis and D. A. I. Goring, J. Pulp Pap. Sci., 1984, 10(5), 139–143 CAS.
- J. M. Willis and D. A. I. Goring, Cellul. Chem. Technol., 1985, 19(6), 679–685 CAS.
- A. M. Emsley and G. C. Stevens, Cellulose, 1994, 1, 26–56 CrossRef CAS.
- M. S. Feather and J. F. Harris, J. Org. Chem., 1965, 30(1), 153–157 CrossRef CAS.
- G. E. Annergren and S. A. Rydholm, Svensk Papperstidning, 1959, 62, 737–746 CAS.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Holzforschung, 2006, 60(2), 162–165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: SEW fractionation of spruce chips. See DOI: 10.1039/c2ra00957a |
| This journal is © The Royal Society of Chemistry 2012 |