Analysis of secondary organic aerosols in air using extractive electrospray ionization mass spectrometry (EESI-MS)†
Lambert A.
Doezema
a,
Teresa
Longin
bc,
William
Cody
b,
Véronique
Perraud
b,
Matthew L.
Dawson
b,
Michael J.
Ezell
b,
John
Greaves
b,
Kathleen R.
Johnson
d and
Barbara J.
Finlayson-Pitts
*b
aDepartment of Chemistry and Biochemistry, Loyola Marymount University, Los Angeles, California, 90045, USA. E-mail: lambert.doezema@lmu.edu; Fax: 310 338 2905; Tel: 310 258 2637
bDepartment of Chemistry, University of California, Irvine, California 92697, USA. E-mail: bjfinlay@uci.edu; Fax: 949 824 2420; Tel: 949 824 7670
cDepartment of Chemistry, University of Redlands, Redlands, California 92373, USA. E-mail: Teresa_longin@redlands.edu; Fax: 909 335-5312; Tel: 909 748 8543
dDepartment of Earth System Science, University of California, Irvine, California 92697, USA. E-mail: Kathleen.johnson@uci.edu; Fax: 949 824 3874; Tel: 949 824 6174
First published on 13th February 2012
Abstract
Extractive electrospray ionization mass spectrometry (EESI-MS) has been shown, in other laboratories, to be a useful technique for the analysis of aerosols from a variety of sources. EESI-MS is applied here, for the first time, to the analysis of secondary organic aerosol (SOA) formed from the reaction of ozone and α-pinene. The results are compared to those obtained using atmospheric pressure chemical ionization mass spectrometry (APCI-MS). The SOA was generated in the laboratory and merged with electrospray droplets. The recovered ions were directed towards the inlet of a triple quadrupole mass spectrometer. Through the use of a denuder to remove gas phase compounds, the EESI-MS technique was found to be effective for measuring the major ozonolysis products either in particles alone or in a combination of vapor phase and particulate products. Due to its relatively simple setup and the avoidance of sample collection and work-up, EESI-MS shows promise as an excellent tool for the characterization of atmospherically relevant particles.
Introduction
Secondary organic aerosol (SOA) is the subject of great interest in the atmospheric chemistry community.1–4 SOA particles are formed from low-volatility oxidation products of organic compounds and are of interest because of their effects on human health and visibility. The light scattering properties of SOA particles and their ability to act as cloud condensation or ice nuclei also means they have a significant impact on climate.5Traditionally, the individual components of SOA have been identified using techniques such as gas chromatography-mass spectrometry and liquid chromatography-mass spectrometry.6–15 However, prior to chromatographic analysis, particles must be collected, extracted, and for a number of compounds, derivatized.12,13,15–19 In addition, these traditional approaches did not detect high molecular weight oligomeric products that were later observed using other methods.20–28
Particle mass spectrometry techniques that use laser ablation or electron impact ionization29–38 provide real-time analysis but often suffer from extensive fragmentation of organic compounds, so that specific molecular mass information and structure cannot be derived from the data. Softer ionization methods such as electrospray ionization (ESI), chemical ionization (CI) and proton-transfer reaction (PTR) combined with mass spectrometry have been used to obtain molecular information on lower molecular mass components of SOA (usually < 300 Da) as well as to detect oligomers up to 2000 Da, but sample collection/concentration of some sort is typically required.20–22,39–45 Atmospheric pressure chemical ionization mass spectrometry (APCI-MS) has been used to make in situ measurements of the gas phase and particle-phase products of a variety of atmospherically relevant reactions such as the oxidation of biogenic terpenes by ozone, NOx, or OH in laboratory studies.46–51 However, the ionization process involves a corona discharge that can induce chemistry in the resulting plasma.
More recently, a number of ambient ionization mass spectrometric techniques have been developed52,53 that can be applied to characterizing particles of atmospheric interest. For example, desorption electrospray ionization mass spectrometry (DESI-MS) has been used by several groups to characterize or quantify organic aerosols.54–57 Bruns et al.27 used atmospheric solids analysis probe mass spectrometry (ASAP-MS) to measure SOA resulting from the ozonolysis of α-pinene and isoprene, the NO3 reaction with α-pinene, and SOA collected from air. However, these techniques require prior collection of the SOA on a substrate.
Extractive electrospray ionization mass spectrometry (EESI-MS) is a recently developed method that involves little sample preparation and has a relatively simple experimental design.58 This technique, which can be carried out on modified, commercially available atmospheric pressure mass spectrometers, involves the interaction of charged electrospray droplets with an aerosol containing the analyte of interest. EESI-MS has been successfully used to study a number of diverse systems including gases and particles from olive oil, aerosolized drugs, human breath, and beer.59–68
We report here the first analysis of SOA using EESI-MS. Of particular importance is the fact that the particles are directly sampled from air with no prior collection onto a substrate. Our studies focus on SOA generated from the reaction of α-pinene with ozone because we have extensive data on the SOA from this reaction using a variety of techniques.27,51,69 In addition, this reaction has been extensively studied in the past because of its atmospheric importance.7,13,20–22,70–76 We compare the results for EESI-MS with those for APCI-MS on the same mass spectrometer in order to provide a comparison for the new technique with a more traditional in situ method.
Experimental
For EESI-MS experiments, SOA was formed in 80 L Teflon chambers from the reaction of α-pinene and ozone (both reactants at 0.7 ppm). The balance of the chamber was filled with air from a zero-air generator (Perma Pure LLC) or with ultra-high purity air (Oxygen Services Company, Ultra Zero Grade). To remove possible contaminants, α-pinene (Sigma Aldrich, 99%+) was purified by column chromatography using aluminum oxide (J. T. Baker, Analyzed Reagent) prior to injection into the chamber by syringe. Ozone was generated by passing a metered flow of ultra-high purity oxygen (Oxygen Services Company, 99.993%) over a low pressure mercury lamp (Jelight Company, 78-2406-1). Ozone concentrations were determined using a photometric ozone analyzer (Teledyne Instruments, Model 400E). Experiments were carried out under dry (no water added) and humid (50% relative humidity) conditions. All EESI-MS data acquisitions were completed within one hour of placing the reactants in the chamber.Typically, two chambers with the same reactant concentrations were prepared. While one chamber was analyzed using EESI-MS, the other chamber was monitored for particle size and concentration using a scanning mobility particle sizer (SMPS, TSI model 3936). SMPS data indicated that during the one hour reaction period the modal particle diameter ranged from approximately 150 nm to 250 nm and the total particle mass ranged from 1500 μg m−3 to 2500 μg m−3.
Measurements were performed on a modified atmospheric pressure ionization (API) triple quadrupole mass spectrometer (Perkin Elmer/AB Sciex API-III) operating in positive ion mode. Fig. 1 depicts the two configurations of the instrument used in this study: EESI mode (Fig. 1a) and the original atmospheric pressure chemical ionization (APCI) mode (Fig. 1b). For EESI-MS spectra, the APCI module was removed, leaving the curtain plate exposed to ambient air. The curtain plate was held at a potential of + 1.0 kV during all experiments. Single quadrupole and MS-MS spectra were acquired using Analyst software.
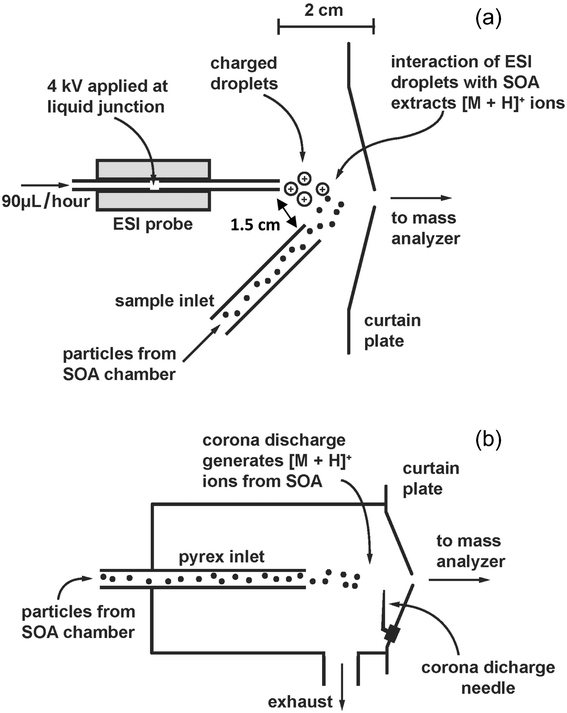 | ||
| Fig. 1 (a) Schematic of the merging of the electrospray droplets and SOA particles in relationship to the curtain plate of the triple quadrupole MS. (b) Schematic of the chamber used for APCI on the same instrument. This chamber and the corona discharge needle are removed when the MS is used in EESI-MS mode. | ||
In the EESI mode, a custom built electrospray source and a ¼′′ Teflon tube for transferring particles from the Teflon chamber to the ionization region were placed near the orifice of the curtain plate. The electrospray tip was placed 2 cm from the curtain plate and aimed slightly off-axis with respect to the orifice. The transfer tube for particles was also placed 2 cm from the orifice but at an angle of approximately 45° with respect to the curtain plate. This system is similar to those used in other studies other than that the electrospray system was operated without a sheath gas for the sake of experimental simplicity.58,59
The custom built electrospray source was constructed using a syringe pump (KDS 100, KD Scientific), perfluoroalkoxyalkane tubing (DuPont), and a 5 cm long fused silica capillary tube with a 100 μm inner diameter (IDEX Health and Science). The solvent tubing and capillary were joined using a stainless steel union (Swagelok). This union created a liquid junction onto which a potential of + 4.0 kV was applied. The EESI solvent was a 50/50 (v/v) mixture of HPLC grade acetonitrile (Sigma-Aldrich) and 0.2% formic acid solution prepared from 18.2 MΩ cm water (Thermo Scientific Barnstead Nanopure) and formic acid (Fisher-Scientific, ACS grade, 88%). The solvent flow was set at a rate of 90 μL h−1.
The aerosol sampling line consisted of 80 cm of ¼′′ stainless steel tubing connected to 20 cm of ¼′′ Teflon tubing that was included to minimize electrostatic interference in the vicinity of the electrospray tip and entrance plate to the mass spectrometer. A 3 cm diameter by 10 cm-long monolith extruded carbon denuder (Novacarb, Mast Carbon Ltd.) operated at room temperature was inserted between the Teflon chamber and the sampling line during some experiments in order to remove gas phase products. This denuder has been shown to remove at least 99% of gas phase reagents and products and transmit more than 92% of the particles formed from the ozonolysis of α-pinene at flow rates of 1.0 L min−1.51 The contents of the Teflon chambers flowed through the sampling line at an approximate rate of 2.5 L min−1 as a result of placing a constant 3 kg mass, that was distributed over a 0.20 m2 area, on top of the chamber. Although this resulted in slightly higher flow rates when the chamber was full, the difference in flow rate for a full chamber versus a half-full chamber was within 15%. SMPS measurements, in which the contents of the Teflon chamber were sampled directly, either through the EESI sample delivery tubing or through the tubing and denuder, indicate that the tubing and denuder had no significant effect on the particle concentration or average diameter.
All sample spectra were corrected for carry-over from previous experiments by subtracting out a spectrum obtained after replacing the reaction chamber with a Teflon chamber filled with ultra high purity air.
For comparison, the APCI-MS spectrum of the reaction of ozone and α-pinene was also acquired using the same instrument in the APCI mode as depicted in Fig. 1b. Ozone and α-pinene were added to a 300 L Teflon chamber to achieve a concentration of 1 ppm. The balance of the chamber was filled with high purity air (Oxygen Services Company, Ultra Zero grade). The mixture of gases and particles in the chamber was passed through a denuder (described above) and through a 22 mm i.d., 33 cm long Pyrex tube at about 1.0 L min−1 into the APCI source of the MS. The source was operated at room temperature.
To provide information on the concentration based response to an individual compound, EESI-MS spectra were acquired for cis-pinonic acid aerosols generated by atomizing solutions of cis-pinonic acid (Aldrich, 98%) in methanol (Aldrich, Optima grade). The solution concentrations were 0.05 mg mL−1, 0.1 mg mL−1, and 0.2 mg mL−1. Each solution was atomized using a constant flow atomizer (TSI model 307601) with nitrogen as the carrier gas (Oxygen Services Company, Ultra High Purity). Particle size distributions were determined using a scanning mobility particle sizer (SMPS, sample flow 0.2 L min−1, sheath flow 2.0 L min−1, impactor 0.071 cm) consisting of an electrostatic classifier (TSI model 3080), a differential mobility analyzer (TSI model 3081) and a condensation particle counter (TSI model 3776). From the SMPS measurements, the mass concentration was obtained assuming a density of 1.2 g cm−3. The number distribution was quite broad and did not capture the tail above ∼500 nm, so that the masses are lower limits.
The mass spectra observed in some experiments contained peaks at m/z 132/134, 122/124, and 63/65 with a 70:30 ratio for each pair. As discussed below, these ions were attributed to copper containing compounds in the laboratory air. The total concentration of copper in the air was estimated by drawing laboratory air through a water bubbler containing 18.2 MΩ cm water at a rate of 1.5 L min−1 for 10–15 h. The air flow was controlled by a mass flow controller (Alicat Scientific) connected to house vacuum. Air was drawn directly into the bubbler through the inlet without coming into contact with tubing or any other substance that might contribute to copper contamination. A procedural blank was prepared by allowing the same volume of 18.2 MΩ cm water to sit in a similar bubbler for 10–15 h without air flowing through it. The concentration of copper in the bubbler water was determined using inductively coupled plasma-mass spectrometry (Nu Instruments AttoM High Resolution ICP-MS). The analysis was conducted in peak-jumping mode using the instrument's unique fast-scan ion optics. Both copper-63 and copper-65 were measured, along with a germanium internal standard to correct for instrumental drift. Concentrations were calculated using a four-point external calibration curve (R2 = 0.9892). Samples were corrected for background copper by subtracting the results of the procedural blank. All glassware was soaked for several hours in 1 M double-distilled nitric acid and rinsed with 18.2 MΩ cm water before use.
Results and discussion
EESI-MS is effective at characterizing the components of the SOA and, in conjunction with a denuder, can differentiate between the analytes in the gas phase and those in particles, as shown in Fig. 2a and 2b. These figures show the EESI-MS spectra between 145 and 190 Da for the SOA generated from the ozonolysis of α-pinene under dry conditions. Table 1 lists the compounds observed in this study. These are some of the most prevalent compounds expected to be present in particles from the ozonolysis of α-pinene based on previous work.7,13,20–22,70–76 The spectrum in Fig. 2a was obtained by placing a denuder between the Teflon reaction chamber and the EESI sample delivery tubing in order to remove gas phase products and excess reactants. Fig. 2b shows a spectrum obtained for the same Teflon reaction chamber without a denuder (i.e. gases plus particles).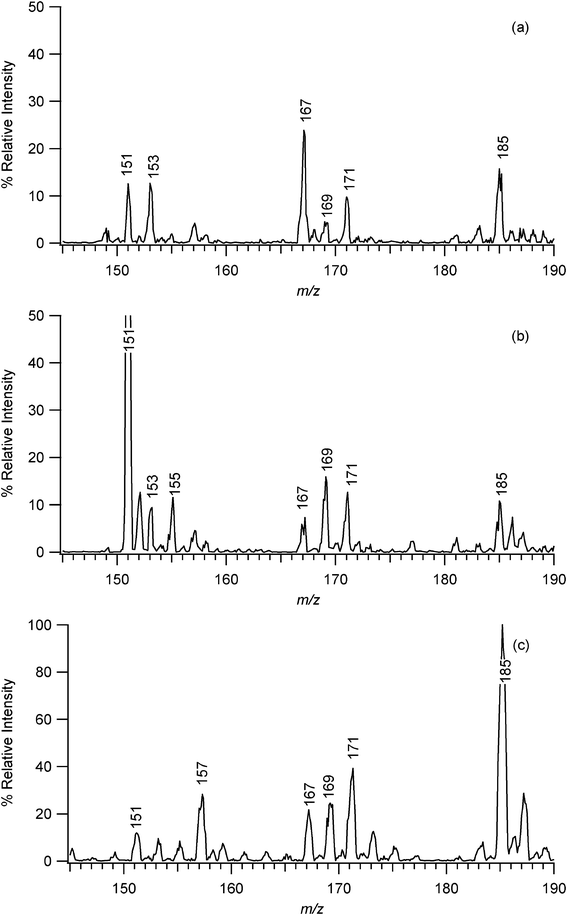 | ||
| Fig. 2 (a) EESI-MS spectrum, corrected for carry-over from previous experiments, of the products of the ozonolysis of α-pinene under dry conditions with a denuder inserted between the Teflon reaction chamber and sample tubing (i.e. particles only). (b) EESI-MS spectrum of the contents of the same reaction chamber without a denuder in place, i.e. gas phase analytes plus particles. Note that the peak at m/z 151 is off-scale; it is actually about 7 times more intense than the other peaks. (c) APCI spectrum of the products of the ozonolysis of α-pinene under dry conditions with a denuder inserted between the Teflon reaction chamber and the APCI inlet. Major peaks are labeled with their respective m/z values. | ||
| Molecular species | Observed m/z | Peak assignment |
|---|---|---|
| Pinic acid (C9H14O4) | 187 | [M + H]+ |
| Pinonic acid (C10H16O3), hydroxypinonaldehydes | 185 | [M + H]+ |
| 167 | [M + H – H2O]+ | |
| Norpinic acid(C8H12O4) | 173 | [M + H]+ |
| Norpinonic acid/pinalic acid (C9H14O3) | 171 | [M + H]+ |
| 153 | [M + H – H2O]+ | |
| Pinonaldehyde (C10H16O2) | 169 | [M + H]+ |
| 151 | [M + H – H2O]+ | |
| 2,2-Dimethylcyclobutane-1-carboxylic acid-3-carboxaldehyde (C8H12O3) | 157 | [M + H]+ |
| Norpinonaldehyde (C9H14O2) | 155 | [M + H]+ |
Peaks at m/z 151, 155, 167 and 169 are common to both spectra but with relative intensities that vary between the sampling with and without the denuder. Both spectra contain peaks of about equal relative intensities at m/z 185, 171, and 153. The peak at m/z 185 is assigned to [M + H]+ while the ion at m/z 167 could correspond to [M + H – H2O]+ of pinonic acid and/or hydroxypinonaldehyde. The peak at m/z 171 could be due to [M + H]+ of norpinonic acid and/or pinalic acid, while the peak at m/z 153 would result from the [M + H – H2O]+ of one or both acids.
The spectrum obtained without a denuder (Fig. 2b) has peaks at m/z 169 and 151 with higher intensities than those in the spectrum obtained using a denuder. These peaks can be attributed to pinonaldehyde. Note that the peak at m/z 151 is off scale on the graph; its intensity is actually about seven times greater than that of the other major peaks. Given the intensity of the [M + H – H2O]+ pinonaldehyde peak, the peak at m/z 152 can be attributed to a 13C isotope ion of pinonaldehyde. The large amount of pinonaldehyde present when not using a denuder relative to that observed when a denuder was employed indicates that most of the pinonaldehyde is present in the gas phase rather than being part of the particles. This is consistent with previous studies of SOA formed from the oxidation of α-pinene75,76 and the relatively high vapor pressure of pinonaldehyde of 1.6 × 10−5 atm at a temperature of 298 K.51,77 The spectrum obtained without a denuder also contains a peak at m/z 155 that is absent from the spectrum obtained with a denuder; this peak is most likely derived from norpinonaldehyde. The data suggest that this ozonolysis product is also primarily in the gas phase rather than being contained in the SOA particles, consistent with its vapor pressure.51
The spectra acquired using EESI-MS are somewhat different from those obtained using APCI-MS. Fig. 2c shows the spectrum obtained using the same mass spectrometer as for the EESI-MS, but in the traditional APCI mode as shown in Fig. 1b. In this case the sample was passed through a denuder before entering the mass spectrometer, so the spectrum in Fig. 2c can be compared directly to the spectrum in Fig. 2a to explore differences in spectrum patterns for particle components. EESI-MS gives major peaks at m/z 185, 171, 167, 153, and 151, while with APCI-MS, the largest peak occurs at m/z 185 with significant peaks at m/z 171, 169, 167, 157 and 151. Similar mass spectra were reported earlier for this reaction when the analysis was done by LC-APCI-MS.6,7
The EESI-MS data show relatively larger [M + H – H2O]+ fragments when compared to their molecular ions. For example, EESI-MS produces a larger peak at m/z 167 [M + H – H2O]+ than at m/z 185 [M + H]+ for pinonic acid/hydroxypinonaldehyde, a larger peak at m/z 151 [M + H – H2O]+ than at m/z 169 [M + H]+ for pinonaldehyde, and a larger peak at m/z 153 [M + H – H2O]+ than at m/z 171 [M + H]+ for norpinonic/pinalic acid. With the APCI-MS technique, the peaks for the molecular ion species dominate for these components. This suggests that, for these particular conditions, APCI-MS is a somewhat “softer” technique than EESI-MS.
EESI-MS and APCI-MS involve different ionization processes. In EESI-MS, it has been reported that differences in analyte solubility between the ESI spray and the sample spray affect the generation of ions.61 This suggests that there is an extractive process that recovers analytes from the sample spray followed by evaporation of the ESI droplets, resulting in the ionization of the component analytes. The electrospray ionization process itself is thought to involve an ion evaporation mechanism in which ions are sputtered from the surface of the highly charged droplets.78 For APCI-MS, samples are drawn directly from the sample chamber through a Pyrex or ceramic tube to the inlet of the mass analyzer. A corona discharge in air in the source of the mass spectrometer causes the formation of ions, and ultimately protonated water clusters [H3O]+ in the positive ion mode. These ions transfer a proton to the analyte molecules if their proton affinity is higher than water.79,80 Although the mechanism of ionization of particles directly by APCI-MS is not known, it is reasonable to suggest that it occurs via uptake of H3O+ or protons at the surface of the SOA, followed by ejection of ions into the gas phase as the charge on the particles builds up. This appears to be a less energetic process than the ejection of ions from the highly charged surface of the electrospray droplets, leading to less fragmentation of the analyte. The fact that the APCI is conducted at room temperature probably contributes to the reduced energy transfer given that when APCI is utilized in LC-MS, temperatures of 250 °C – 600 °C are typically used to evaporate the solvent and fragmentation is more extensive than occurs for ESI.
Mass spectra for SOA generated in the presence of water vapor were similar to those for the dry SOA, except that they exhibited more ions in the higher (> 300 Da) mass range. Fig. 3a shows the EESI-MS for ozonolysis of α-pinene at 50% relative humidity up to 500 Da, while Fig. 3b shows the results for the reaction under dry conditions. Similar higher molecular weight products have been observed in other SOA studies and have been attributed to oligomeric products.20–25,27,74 One possibility is that these higher molecular weight peaks are due to adducts formed during the electrospray droplet evaporation and ionization process. However, that seems unlikely based on studies by Reinhardt et al.21 in which they explored the high molecular weight products formed from the ozonolysis of α-pinene using Fourier transform ion cyclotron resonance mass spectrometry with an ESI source. They looked at the effect of source CID voltage on the observed spectra since noncovalent adducts should be disrupted at higher CID voltages. Since their spectra did not change as a function of source CID voltage, they concluded that the higher molecular weight compounds must be oligomers rather than noncovalent adducts. In addition, Gao et al.81 used ESI-MS to study the ozonolysis of α-pinene in a flow tube reactor. They observed a species they attributed to a dimer and used LC-MS to confirm that the dimer was not an artifact of the ESI analysis. Given that EESI-MS ultimately involves the same solvent evaporation and ionization process as more conventional ESI-MS, it is likely that the higher molecular weight products seen in this study are also oligomers rather than noncovalent adducts. Finally, high molecular weight signals attributed to oligomers have been seen by ionization techniques other than ESI, also suggesting that these peaks are not merely adducts of the ESI process.20,23,24,27
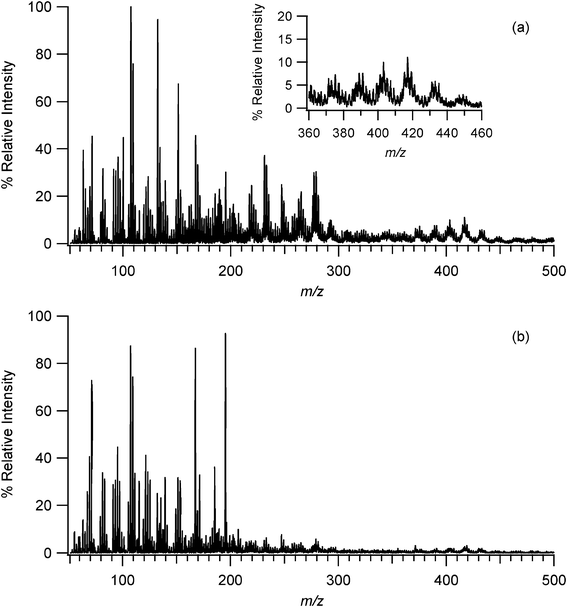 | ||
| Fig. 3 (a) EESI-MS of SOA generated from the ozonolysis of α-pinene at 50% relative humidity. Inset shows the presence of oligomers at higher molecular weights. (b) EESI-MS of SOA generated under dry conditions showing reduced numbers of ions in the m/z 200–500 region. | ||
In terms of the analytical technique, detection of these higher mass compounds demonstrates that EESI-MS is capable of analyzing the components of particles of atmospheric importance throughout a large mass range.
Spectra obtained by EESI-MS are expected to be subject to similar matrix and solvent effects as traditional ESI, and to have response factors that depend on the particular analyte. This can be seen, for example, in the spectra in Fig. 2 where the [M + H]+ peak due to pinic acid at m/z 187 is small relative to that at m/z 185 due to pinonic acid/hydroxypinonaldehyde, yet pinic acid is known to be a major component of SOA from α-pinene ozonolysis.7,13 This is due the larger proton affinities of pinonic acid and hydroxypinonaldehydes which both contain a ketone group, compared to pinic acid which only contains –COOH groups that do not protonate.6,7
The EESI-MS of cis-pinonic acid particles was obtained by sampling an atomized solution of cis-pinonic acid in methanol in order to estimate a limit of detection for a component of the SOA. Peaks at m/z 185 (assigned to [M + H]+) and m/z 167 (assigned to [M + H – H2O]+) were observed as expected; the mass spectrum from m/z 145–190 Da for the 0.1 mg mL−1 solution is provided in the Supplementary Information (Fig. S1†). The signal intensities increased linearly with particle mass within the uncertainty of the SMPS measurements, indicating that at least for pure compounds, this method should be quantitative. As an example, using this linear relationship for the peak at m/z 167 and the intensity of the noise in this region, the EESI-MS detection limit for cis-pinonic acid is estimated to be ∼20 μg m−3 under these conditions. However, the impact of matrix effects where cis-pinonic acid is in a complex mixture with other species needs to be investigated. Nevertheless, this detection limit could be improved through the use of increased integration times, optimized geometry and potentially using different solvent systems in which cis-pinonic acid has a higher solubility since a previous report indicated that EESI-MS sensitivity increases as the solubility of the analyte in the EESI solvent increases.61
An example of the influence of matrix factors in this particular system is the sporadic detection of copper compounds in laboratory air at concentrations that were independently estimated by ICP-MS to be 500–600 ng m−3 (see Supplementary Information†). Peaks would appear at m/z 132 and 134 that varied in strength depending on the day and time of day (Fig. 4). Smaller peaks at m/z 63 and 65 correlated very well with the presence of the m/z 132 and 134 peaks, and both sets of peaks exhibited a constant intensity ratio of approximately 70:30 as expected for copper isotopes. Peaks at m/z 122 and 124 showed a similar ratio. Tandem mass spectra (MS-MS) of m/z 132 and 122 further supported the conclusion that the peaks were due to copper adducts as described in the Supplementary Information.†
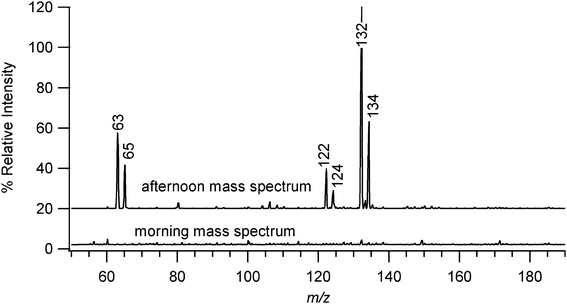 | ||
| Fig. 4 EESI-MS spectra of the mass region that typically contained peaks resulting from sporadic copper contamination of the laboratory air. Major peaks are labeled with their respective m/z values. Spectra are vertically offset from the baseline for the sake of clarity. | ||
A number of tests indicated that the copper was in the laboratory air, and formed a complex with the acetonitrile solvent (see Supplementary Information†). For example, the copper peaks were significantly reduced in intensity if formic acid was not included in the acetonitrile–water solvent mixture and were completely absent when methanol was used as a solvent. It appears that acetonitrile is essential for the extraction of copper from the air and the presence of acid greatly increases the extraction or ionization efficiency. The estimated detection limit for copper in the form of this complex in the 50/50 acetonitrile/0.2% formic acid solvent is 3 ng m−3, assuming that all of the copper is present as the complex and that the signal is proportional to mass, as is the case for pinonic acid.
Conclusions
EESI-MS has been applied to analyzing particles of atmospheric relevance. This is a potentially useful technique that can be carried out with minimal modifications of existing API mass spectrometers. Additionally, the technique does not require prior collection of particles on a substrate and thus can be used to obtain real-time measurements. Compared to many existing techniques, EESI-MS provides soft ionization that facilitates identification of different compounds in a complex mixture such as SOA. In this study, the EESI process involves the interaction of ESI droplets with non-solvated SOA particles. The droplets must therefore be dissolving components from these particles. The fact that this technique produces spectra similar to those obtained by APCI-MS, a technique with which the major known products of α-pinene ozonolysis including oligomers have been identified, indicates that this is an effective method for generating ions from particles. Future studies will focus on optimization of the technique, including the development of authentic, atmospherically relevant standards to determine limits of detection for SOA components individually and in mixtures, determining the recovery efficiency, the impact of solvent composition, investigation of the use of the negative ion mode for carboxylic acids, the use of a sheath gas around the solvent spray, and the use of nano-EESI-MS which has been shown to enhance sensitivity.82Acknowledgements
We are grateful to the National Science Foundation (grant #0909227) for support of this work. We also thank Sergey Nizkorodov for advice on building the electrospray apparatus and James N. Pitts for helpful discussions and comments on the manuscript.References
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CAS.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Atmos. Chem. Phys., 2005, 5, 1053–1123 CAS.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS.
- IPCC, Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2007 Search PubMed.
- M. Glasius, M. Duane and B. R. Larsen, J. Chromatogr., A, 1999, 833, 121–135 CrossRef CAS.
- R. Winterhalter, R. Van Dingenen, B. R. Larsen, N. R. Jensen and J. Hjorth, Atmos. Chem. Phys. Discuss., 2003, 3, 1–39 CrossRef.
- J. C. Chow, P. Doraiswamy, J. G. Watson, L. W. A. Chen, S. S. H. Ho and D. A. Sodeman, J. Air Waste Manage. Assoc., 2008, 58, 141–163 Search PubMed.
- E. A. Stone, C. J. Hedman, J. B. Zhou, M. Mieritz and J. J. Schauer, Atmos. Environ., 2010, 44, 312–319 Search PubMed.
- B. J. Williams, A. H. Goldstein, N. M. Kreisberg and S. V. Hering, Aerosol Sci. Technol., 2006, 40, 627–638 Search PubMed.
- Z. Chowdhury, M. Zheng, J. J. Schauer, R. J. Sheesley, L. G. Salmon, G. R. Cass and A. G. Russell, J. Geophys. Res., 2007, 112, DOI:10.1029/2007JD008386.
- M. Jaoui and R. M. Kamens, J. Geophys. Res., 2001, 106, 12541–12558 Search PubMed.
- J. Yu, D. R. Cocker III, R. J. Griffin, R. C. Flagan and J. H. Seinfeld, J. Atmos. Chem., 1999, 34, 207–258 CrossRef CAS.
- B. R. Larsen, D. DiBella, M. Glausius, R. Winterhalter, N. R. Jensen and J. Hjorth, J. Atmos. Chem., 2001, 38, 231–276 CrossRef CAS.
- T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, E. O. Edney, M. Jaoui, M. Zheng, X. A. Ding and E. S. Edgerton, J. Air Waste Manage. Assoc., 2010, 60, 1388–1399 Search PubMed.
- J. Yu, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 1998, 32, 2357–2370 CrossRef.
- R. Szmigielski, J. D. Surratt, R. Vermeylen, K. Szmigielska, J. H. Kroll, N. L. Ng, S. M. Murphy, A. Sorooshian, J. H. Seinfeld and M. Claeys, J. Mass Spectrom., 2007, 42, 101–116 CrossRef CAS.
- M. Claeys, R. Szmigielski, I. Kourtchev, P. vanderVeken, W. Maenhaut, M. Jaoui, T. E. Kleindienst, M. Lewandowski, H. H. Offenberg and E. O. Edney, Environ. Sci. Technol., 2007, 41, 1628–1634 CrossRef CAS.
- M. Jaoui, T. E. Kleindienst, M. Lewandowski and E. O. Edney, Anal. Chem., 2004, 76, 4765–4778 CrossRef CAS.
- M. P. Tolocka, M. Jang, J. M. Ginter, F. J. Cox, R. M. Kamens and M. V. Johnston, Environ. Sci. Technol., 2004, 38, 1428–1434 CrossRef CAS.
- A. Reinhardt, C. Emmenegger, B. Gerrits, C. Panse, J. Dommen, U. Baltensperger, R. Zenobi and M. Kalberer, Anal. Chem., 2007, 79, 4074–4082 CrossRef CAS.
- S. Gao, M. Keywood, N. L. Ng, J. Surratt, V. Varutbangkul, R. Bahreini, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem. A, 2004, 108, 10147–10164 CrossRef CAS.
- M. Kalberer, M. Sax and V. Samburova, Environ. Sci. Technol., 2006, 40, 5917–5922 CrossRef CAS.
- M. Kalberer, D. Paulsen, M. Sax, M. Steinbacher, J. Dommen, A. S. H. Prevot, R. Fisseha, E. Weingartner, V. Frankevich, R. Zenobi and U. Baltensperger, Science, 2004, 303, 1659–1662 CrossRef CAS.
- K. A. Denkenberger, R. C. Moffet, J. C. Holecek, T. P. Rebotier and K. A. Prather, Environ. Sci. Technol., 2007, 41, 5439–5446 CrossRef CAS.
- S. Gao, N. L. Ng, M. Keywood, V. Varutbangkul, R. Bahreini, A. Nenes, J. He, K. Y. Yoo, J. L. Beauchamp, R. P. Hodyss, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2004, 38, 6582–6589 CrossRef CAS.
- E. A. Bruns, V. Perraud, J. Greaves and B. J. Finlayson-Pitts, Anal. Chem., 2010, 82, 5922–5927 CrossRef CAS.
- J. Dommen, A. Metzger, J. Duplissy, M. Kalberer, M. R. Alfarra, A. Gascho, E. Weingartner, A. S. H. Prevot, B. Verheggen and U. Baltensperger, Geophys. Res. Lett., 2006, 33, L13805 Search PubMed.
- S. Wang, C. A. Zordan and M. V. Johnston, Anal. Chem., 2006, 78, 1750–1754 CrossRef CAS.
- D. A. Lake, M. P. Tolocka, M. V. Johnston and A. S. Wexler, Environ. Sci. Technol., 2003, 37, 3268–3274 CrossRef CAS.
- B. Oktem, M. P. Tolocka and M. V. Johnston, Anal. Chem., 2004, 76, 253–261 CrossRef.
- A. Zelenyuk and D. Imre, Aerosol Sci. Technol., 2005, 39, 554–568 CAS.
- A. Zelenyuk, J. Yang, E. Choi and D. Imre, Aerosol Sci. Technol., 2009, 43, 411–424 CAS.
- K. A. Prather, C. D. Hatch and V. H. Grassian, Annu. Rev. Anal. Chem., 2008, 1, 485–514 Search PubMed.
- D. M. Murphy, D. J. Cziczo, K. D. Froyd, P. K. Hudson, B. M. Matthew, A. M. Middlebrook, R. E. Peltier, A. Sullivan, D. S. Thomson and R. J. Weber, J. Geophys. Res., 2006, 111, DOI:10.1029/2006JD007340.
- A. M. Middlebrook, D. M. Murphy, S. H. Lee, D. S. Thomson, K. A. Prather, R. J. Wenzel, D. Y. Liu, D. J. Phares, K. P. Rhoads, A. S. Wexler, M. V. Johnston, J. L. Jimenez, J. T. Jayne, D. R. Worsnop, I. Yourshaw, J. H. Seinfeld and R. C. Flagan, J. Geophys. Res., 2003, 108, DOI:10.1029/2001JD000660.
- M. R. Canagaratna, J. T. Jayne, J. L. Jimenez, J. D. Allan, M. R. Alfarra, Q. Zhang, T. B. Onasch, F. Drewnick, H. Coe, A. Middlebrook, A. Delia, L. R. Williams, A. M. Trimborn, M. J. Northway, P. F. DeCarlo, C. E. Kolb, P. Davidovits and D. R. Worsnop, Mass Spectrom. Rev., 2007, 26, 185–222 CrossRef CAS.
- J. L. Jimenez, J. T. Jayne, Q. Shi, C. E. Kolb, D. R. Worsnop, I. Yourshaw, J. H. Seinfeld, R. C. Flagan, X. F. Zhang, K. A. Smith, J. W. Morris and P. Davidovits, J. Geophys. Res., 2003, 108, DOI:10.1029/2001JD001213.
- A. P. Bateman, M. L. Walser, Y. Desyaterik, J. Laskin, A. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2008, 42, 7341–7346 CrossRef CAS.
- J. N. Smith, K. F. Moore, P. H. McMurry and F. L. Eisele, Aerosol Sci. Technol., 2004, 38, 100–110 CAS.
- L. Wang, A. F. Khalizov, J. Zheng, W. Xu, Y. Ma, V. Lal and R. Y. Zhang, Nat. Geosci., 2010, 3, 238–242 Search PubMed.
- R. Holzinger, J. Williams, F. Herrmann, J. Lelieveld, N. M. Donahue and T. Rockmann, Atmos. Chem. Phys., 2010, 10, 2257–2267 Search PubMed.
- T. Thornberry, D. M. Murphy, D. S. Thomson, J. de Gouw, C. Warneke, T. S. Bates, P. K. Quinn and D. Coffman, Aerosol Sci. Technol., 2009, 43, 486–501 Search PubMed.
- R. L. N. Yatavelli and J. A. Thornton, Aerosol Sci. Technol., 2010, 44, 61–74 Search PubMed.
- H. J. Tobias and P. J. Ziemann, Environ. Sci. Technol., 2000, 34, 2105–2115 CrossRef CAS.
- S. M. Aschmann, A. Reissel, R. Atkinson and J. Arey, J. Geophys. Res., 1998, 103, 25553–25561 CrossRef CAS.
- T. Hoffmann, R. Bandur, U. Marggraf and M. Linscheid, J. Geophys. Res., 1998, 103, 25569–25578 CrossRef CAS.
- B. Warscheid and T. Hoffmann, Rapid Commun. Mass Spectrom., 2001, 15, 2259–2272 CrossRef CAS.
- B. Warscheid and T. Hoffmann, Atmos. Environ., 2001, 35, 2927–2940 CrossRef CAS.
- S. M. Aschmann, R. Atkinson and J. Arey, J. Geophys. Res., 2002, 107, DOI:10.1029/2001JD001234.
- V. Perraud, E. A. Bruns, M. J. Ezell, S. N. Johnson, J. Greaves and B. J. Finlayson-Pitts, Environ. Sci. Technol., 2010, 44, 5887–5893 CrossRef CAS.
- D. R. Ifa, C. P. Wu, Z. Ouyang and R. G. Cooks, Analyst, 2010, 135, 669–681 RSC.
- A. Venter, M. Nefliu and R. G. Cooks, TrAC, Trends Anal. Chem., 2008, 27, 284–290 CrossRef CAS.
- H. Chen, M. Li, Y. P. Zhang, X. Yang, J. J. Lian and J. M. Chen, J. Am. Soc. Mass Spectrom., 2008, 19, 450–454 CrossRef CAS.
- M. Li, H. Chen, X. Yang, J. Chen and C. Li, Atmos. Environ., 2009, 43, 2717–2720 CrossRef CAS.
- J. Laskin, A. Laskin, P. J. Roach, G. W. Slysz, G. A. Anderson, S. A. Nizkorodov, D. L. Bones and L. Q. Nguyen, Anal. Chem., 2010, 82, 2048–2058 CrossRef CAS.
- M. Li, H. Chen, B. F. Wang, X. Yang, J. J. Lian and J. M. Chen, Int. J. Mass Spectrom., 2009, 281, 31–36 Search PubMed.
- H. Chen, A. Venter and R. G. Cooks, Chem. Commun., 2006, 2042–2044 RSC.
- J. Dong, Y. H. Rezenom and K. K. Murray, Rapid Commun. Mass Spectrom., 2007, 21, 3995–4000 CrossRef.
- C. A. Marquez, H. Wang, F. Fabbretti and J. O. Metzger, J. Am. Chem. Soc., 2008, 130, 17208–17209 CrossRef CAS.
- W. S. Law, R. Wang, B. Hu, C. Berchtold, L. Meier, H. W. Chen and R. Zenobi, Anal. Chem., 2010, 82, 4494–4500 CrossRef CAS.
- W. S. Law, H. W. Chen, R. Balabin, C. Berchtold, L. Meier and R. Zenobi, Analyst, 2010, 135, 773–778 RSC.
- L. A. Zhu, Z. Hu, G. Gamez, W. S. Law, H. W. Chen, S. P. Yang, K. Chingin, R. M. Balabin, R. Wang, T. T. Zhang and R. Zenobi, Anal. Bioanal. Chem., 2010, 398, 405–413 Search PubMed.
- H. Chen, A. Wortmann, W. Zhang and R. Zenobi, Angew. Chem., Int. Ed., 2007, 46, 580–583 CrossRef CAS.
- C. Y. Lee and J. Shiea, Anal. Chem., 1998, 70, 2757–2761 CrossRef CAS.
- G. Gamez, L. A. Zhu, A. Disko, H. W. Chen, V. Azov, K. Chingin, G. Kramer and R. Zenobi, Chem. Commun., 2011, 47, 4884–4886 RSC.
- J. H. Ding, S. P. Yang, D. P. Liang, H. W. Chen, Z. Z. Wu, L. L. Zhang and Y. L. Ren, Analyst, 2009, 134, 2040–2050 RSC.
- H. W. Gu, B. Hu, J. Q. Li, S. P. Yang, J. Han and H. W. Chen, Analyst, 2010, 135, 1259–1267 RSC.
- V. Perraud, E. A. Bruns, M. J. Ezell, S. N. Johnson, Y. Yu, M. L. Alexander, A. Zelenyuk, D. Imre, W. L. Chang, D. Dabdub, J. F. Pankow and B. J. Finlayson-Pitts, PNAS, 2012, DOI:10.1073/pnas.1119909109.
- M. Jang and R. M. Kamens, Atmos. Environ., 1999, 33, 459–474 CrossRef CAS.
- M. Jaoui and R. M. Kamens, J. Atmos. Chem., 2003, 44, 259–297 Search PubMed.
- R. Bahreini, M. D. Keywood, N. L. Ng, V. Varutbangkul, S. Gao, R. C. Flagan, J. H. Seinfeld, D. R. Worsnop and J. L. Jimenez, Environ. Sci. Technol., 2005, 39, 5674–5688 CrossRef CAS.
- A. Lee, A. H. Goldstein, M. D. Keywood, S. Gao, V. Varutbangkul, R. Bahreini, N. L. Ng, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res., 2006, 111, DOI:10.1019/2005JD006437.
- W. A. Hall and M. V. Johnston, Aerosol Sci. Technol., 2011, 45, 37–45 Search PubMed.
- R. Atkinson and J. Arey, Atmos. Environ., 2003, 37, S197–S219 CAS.
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- J. F. Pankow and W. E. Asher, Atmos. Chem. Phys., 2008, 8, 2773–2796 CAS.
- S. Nguyen and J. B. Fenn, PNAS, 2007, 104, 1111–1117 CrossRef CAS.
- C. W. Spicer, D. V. Kenny, W. J. Shaw, K. M. Busness and E. G. Chapman, Environ. Sci. Technol., 1994, 28, 412–420 Search PubMed.
- C. J. Proctor and J. F. J. Todd, Org. Mass Spectrom., 1983, 18, 509–516 CrossRef CAS.
- Y. Gao, W. A. Hall and M. V. Johnston, Environ. Sci. Technol., 2010, 44, 7897–7902 CrossRef CAS.
- J. Dong, Ph. D. Thesis “Merged electrospray ionization mass spectrometry”, Louisiana State University, 2009 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra00961g/ |
| This journal is © The Royal Society of Chemistry 2012 |