Metal-enhanced fluorescence of carbon dots adsorbed Ag@SiO2 core-shell nanoparticles
Cuiyan
Li
,
Yihua
Zhu
*,
Xiaoqing
Zhang
,
Xiaoling
Yang
and
Chunzhong
Li
Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: yhzhu@ecust.edu.cn; Fax: +86 21 6425 0624; Tel: +86 21 6425 2022
First published on 13th January 2012
Abstract
Metal-enhanced fluorescence carbon dots adsorbed Ag@SiO2 composite nanoparticles, where the silica-shell is used to control the distance between silver-core and fluorescent carbon dots (CDs). A more than four-fold increase of fluorescence intensity of CDs was obtained, besides, a largely enhanced upconversion property was also observed.
Recently, fluorescent nanoparticles, primarily semiconductor quantum dots (QDs), have attracted much attention for a variety of purposes and applications in biology and medicine, for example, in immuno-labeling,1 as cell markers,2 in cell motility assays,3 in biological assembly,4 and especially in optical imaging agents,5 due to their photostability and other excellent optical properties. Semiconductor QDs (such as ZnS, CdS, CdSe) and the related core-shell nanocrystals (like CdSe/ZnS) are usually used. However, the release of Cd2+ ions arouse cytotoxicity and is a potential environmental hazard, which limits the application of quantum dots.6 Due to the urgent need of environmentally-friendly fluorescent nanoparticles, the emerging carbon dots (CDs) appear to be a promising alternative to semiconductor QDs in many applications such as bioimaging, disease detection, and drug delivery7 because of their superiority in chemical inertness, biocompatibility, low toxicity, cheaper cost and promising upconversion property.8
However, the fluorescent intensity of CDs is relatively low in most reports,9 but metal (such as gold or silver) has an enhanced effect on the fluorophore, as known as metal-enhanced fluorescence (MEF).10 It is well established that the interactions of fluorophores with silver nanoparticles results in an increased photostability, fluorescence enhancement, and a decreased lifetime due to increased rates of system radioactive decay.11 The fluorescence enhancement noted in these studies can stem from a mechanism of surface plasmon resonance.12 Excellent overlap of the CDs absorption/emission spectrum with the scattering spectrum of the silver surface is required for effective surface plasmon resonance, while CDs show a broad absorption and emission spectrum, which provides a good overlap with the silver scattering spectrum.13
The spontaneous emission of light by molecules and atoms at nanostructured metallic surfaces is remarkably modified due to a complex interplay of enhancing and quenching physicochemical processes. Fluorescence enhancement can be promoted by surface plasmons excited in metal and by a modified density of photon states in a nanostructured surface,14 while quenching processes include chemical bonding and nonradiative energy transfer from the luminescent species to the metal. Importantly, the degree of metal-fluorophore interactions strongly depends on the distance between the fluorophore and the metal surface. There are various methods established to control this distance. Here we chose a controllable silica shell around the silver core to achieve this goal with several advantages: firstly, silica layers offer the chemical inertness, and offer the versatility needed for the conjugation of biomolecules or fluorophores; secondly, silica shell protects the silver core from ions present in biological media; thirdly, silica layers allow for the distance dependent MEF phenomenon. Besides, regarding the silica-shell and CDs are both negatively charged on their surface, a positively charged polyelectrolyte PAH, poly(allylamine hydrochloride) is used to adsorb Ag@SiO2 nanoparticles and CDs by layer-by-layer (LbL) electrostatic assembly approach.
Herein, our concentric multilayer architecture features a silver core on which is grown a silica spacer shell with a controlled and uniform thickness to prevent quenching of fluorescent CDs by the metal at very close range. As represented in Fig. 1, the preparation of the composite nanoparticles was undertaken in four steps: (1) Ag colloids are prepared by reduction of silver nitrate by PVP; (2) deposit a controllable silica layer onto the Ag colloids to obtain Ag@SiO2 core-shell nanoparticles (A, B and C present the different silica-shell thicknesses, respectively); (3) modify the surface of the silica with positively charged PAH; (4) hybrid assembly between positively charged Ag@SiO2 nanoparticles and negatively charged CDs by electrostatic interaction.
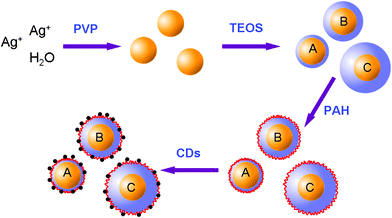 | ||
| Fig. 1 Schematic representation of the preparation of the CDs-adsorbed Ag@SiO2 composite nanoparticles. | ||
With the previous method in our group,15 the hydrophilic CDs were prepared by a novel and facile approach using an impregnation method, with mesoporous silica (MS) spheres as the nanoreactors and citric acid as the carbon precursor. The as-prepared CDs with a high photoluminescence (PL) efficiency of about 23% were well dispersed in water with sizes of 2 ± 0.5 nm. The CDs which give off blue luminescence also show excellent upconversion luminescence properties. This upconverted PL property of CDs could be attributed to a multiphoton active process similar to previous reports of CDs,7,16 which suggested that CDs may be used as a powerful energy-transfer component in photocatalyst design for application in environmental and energy issues.
Silver nanoparticles were prepared through a modified polyol process.17 Typically, 0.75 g polyvinylpyrrolidone (PVP-K45) was dissolved in 75 mL ethanol at room temperature, then 0.05 g AgNO3 was added and stirred for 30 min. After complete dissolution of AgNO3 and the solution was transparent again, the system was heated to 120 °C and the reaction proceeded for 2 h. At the end of the reaction, the colloidal dispersion was cooled with water until the system reached room temperature. The silver colloid can be separated easily from the ethylene glycol after the addition of a large amount of acetone (500 mL) by ultraphonic and centrifugation at the rate of 9000 rad min−1, and the volume was adjusted to 100 mL dispersed in anhydrous ethanol.
To deposit a silica shell, we used a modified version of the procedure reported by Graf et al.18 5 mL synthesized silver nanoparticles were redispersed in 45 mL ethanol with 1 mL ammonia, and to this solution a various amount of tetraethyl orthosilicate (TEOS) solution (10 mM in ethanol) was added (from 0.5 to 10 mL) to obtain different thickness of silica shell. After centrifuging and washing with ethanol, the Ag@SiO2 nanoparticles were redispersed in 20 mL ethanol, then 0.5 mL PAH (1%) was added to function as the surface of positive charge. At last, the surface-modified particles were dispersed in CDs solution at the volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and stirred at room temperature overnight. The carbon QDs were attached to the surface of the functionalized silica surfaces through electrostatic interaction, resulting in the targeted CDs-adsorbed Ag@SiO2 composite particles were acquired. All the glass apparatus were immersed in aqua regia solution for 24 h before being used.
:1 and stirred at room temperature overnight. The carbon QDs were attached to the surface of the functionalized silica surfaces through electrostatic interaction, resulting in the targeted CDs-adsorbed Ag@SiO2 composite particles were acquired. All the glass apparatus were immersed in aqua regia solution for 24 h before being used.
Fig. 2 (a) indicates the prepared PVP-stabilized silver nanoparticles have an average size of 50 ± 5 nm. It is known that the size of the metal core can have an influence on MEF, and it has been reported that the enhancement efficiency of MEF for fluorophores near the surface of silver nanoparticles reaches a maximum for particle size around 50 nm and remains relatively constant in the 40–70 nm range,19 therefore we can assume that MEF efficiency will be relatively uniform across the span of our core-shell nanoparticles. Then, uniform silica spacer shells were grown onto the silver nanoparticles as certified in Fig. 2 (b). The amount of TEOS used in the synthesis was adjusted to vary the thickness of the spacer shell in order to optimize the fluorescence enhancement. The thickness of the silica layers were 10 ± 2 nm (Fig. 2 c), 20 ± 2 nm (Fig. 2 d) and 30 ± 2 nm (Fig. 2 e), respectively.
 | ||
| Fig. 2 (a) SEM photographs of Ag colloids. (b) SEM photographs of Ag@SiO2 core-shell nanoparticles. (c–e) TEM images of Ag@SiO2 core-shell nanoparticles, where the silica shell thickness is 10, 20 and 30 nm, respectively. | ||
As shown in Fig. 3, the surface of silver colloids with PVP coating was positively charged (zeta-potential = 9.8 mV) while the surface of silica-coated silver nanoparticles was negatively charged (zeta potential = −20.0 mV) at pH 7.8 because of the silane group on the silica shell. The surface of the PAH modified silica-coated silver nanoparticles was positively charged (zeta potential = 10.4 mV) at pH 7.8, which was because the alkylamines exist predominantly as positively charged R–NH3+ groups.20 Since the electrostatic interaction acts as the driving force for the assembly of CDs onto amino modified nanoparticles, the targeted enhancing luminescent CDs-adsorbed Ag@SiO2 composite nanoparticles (CNPs) with the negatively charged surface (zeta potential = −12.8 mV) at pH 7.8 was successfully prepared. This charge originated from the ionization of the carboxylic acid and hydroxy groups that were located on the surface of CDs.21 It can be deduced that CDs were well-separated from each other on the silica surface, and the attachment was sufficiently strong that the CDs remained attached to the silica shell when the nanoparticles were centrifuged out of a solution and redispersed.
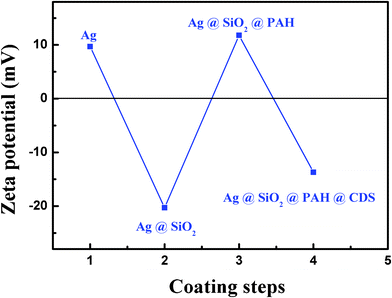 | ||
| Fig. 3 Zeta potential traced with every step of the process. | ||
Fig. 4 shows the dispersion of Ag@SiO2 nanoparticles after the particles were prepared and kept in ethanol for one month. It can be seen that the Ag@SiO2 particles were well dispersed without any aggregation, except a few Ag-free silica spheres which were hard to avoid totally; while the structure and dispersion are still maintained without any damage or deformation after one month, which presents the good stability of the as-prepared nanoparticles structure.
 | ||
| Fig. 4 TEM image of Ag@SiO2 core-shell nanoparticles with the shell thickness of 20 nm (CNP-B). | ||
Fig. 5 (a) shows the fluorescence emission intensity of CDs in ethanol and in CNPs with different silica-shell thickness. For the shell thickness of 10 nm (CNPs-A for short), the fluorescence emission intensity of CDs has a small decline, as a result of quenching due to the too close distance from the metal surface. For the shell thickness of 20 nm (CNPs-B), the fluorescence emission intensity of CDs has a dramatic increase of almost 4-fold, owing to the metal-enhancement. While for shell thickness of 30 nm (CNPs-C), the fluorescence emission intensity of CDs drops by a half, because the silica-shell is too thick to make the silver-core affect fluorescent CDs. Thus, in this work, 20 nm spacer thickness is the best distance for metal-enhancement fluorescence. Though the intrinsic mechanism of CD luminescence is not yet fully understood, the metal-enhancement fluorescence was confirmed to be still applied when CDs were used as fluorophores. Although many papers report nanoballs by etching the metal-core as a comparison, we think this comparison method is not scientifically accurate when the nanoparticles were diluted by adding etchant, and are washed away by incomplete centrifugation and redispersion. Therefore, we simply compared the fluorescent intensity of CDs with that of the composited nanoparticles.
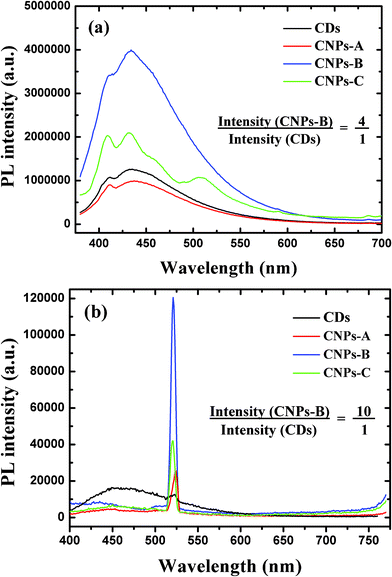 | ||
| Fig. 5 (a) Fluorescence emission intensity of CDs in ethanol and in CNPs with the silica-shell thickness of 10 nm, 20 nm and 30 nm, respectively (all volume ratio was 1:1). (b) Upconverted PL properties of CDs in ethanol and in CNPs with the silica-shell thickness of 10 nm, 20 nm and 30 nm, respectively, at excitation wavelength 780 nm (all volume ratio was 1:1). | ||
Fig. 5 (b) shows the upconverted emission spectra of CDs in ethanol and in CNPs with different silica-shell thickness. Compared with the cyan line (wavelength 780 nm) in Fig. 1(b), when CDs were dispersed in the CNPs solutions, its upconversion changed a lot: the PL peak at around 450 nm was extremely decreased to nearly invisible, while the small sharp peak at 520 nm was largely enhanced. For the shell thickness of 10 nm (CNPs-A), the upconverted emission intensity of CDs has climbed to approximately twice that of CDs in ethonal, even though the fluorescence emission intensity is lower. For the shell thickness of 20 nm (CNPs-B), the upconverted emission intensity of CDs in CNPs was 10-fold higher than CDs in ethanol. While for shell thickness of 30 nm (CNPs-C), the upconverted emission intensity of CDs drops back. So, 20 nm spacer thickness is also the best distance for metal-enhancement upconverted PL properties. Although the explicit principle for the intensely enhancement upconverted PL is not clear up to now, we try to explain it from multiphotons theory and energy transfer theory, since the PL intensity at 450 nm decreases obviously while the intensity at 520 nm enormously increases.
In conclusion, we have investigated the MEF effect of CDs-adsorbed Ag@SiO2 nanocomposites, where the silica-spacer thickness is adjusted to optimize the metal-fluorophores distance and the fluorescence enhancement. A more than four-fold increase of fluorescence intensity of CDs was obtained, besides, a largely enhanced upconversion property was also observed, we try to explain it from multiphotons theory and energy transfer theory, since the PL intensity at 450 nm decreases obviously while the primary small peak at 520 nm enormously increases. Further experiments with other types of metal or alloy nanostructures will allow the clarification of the details of QDs fluorescent properties.
This work was supported by the National Natural Science Foundation of China (20925621, 20976054, and 21176083), the Fundamental Research Funds for the Central Universities, the Program for Changjiang Scholars and Innovative Research Team in University (IRT0825), and the Shanghai Leading Academic Discipline Project (project number: B502) for financial supports.
References
- F. Osaki, T. Kanamori, S. Sando, T. Sera and Y. A. Aoyagi, J. Am. Chem. Soc., 2004, 126, 6520 CrossRef CAS.
- H. Y. Xie, C. Zuo, Y. Liu, Z. L. Zhang, D. W. Pang, X. L. Li, J. P. Gong, C. Dickinson and W. Zhou, Small, 2005, 1, 506 CrossRef CAS.
- W. J. Park, R. Boudreau, M. Le Gros, D. Gerion, D. Zanchet, C. M. Micheel, S. C. Williams, A. P. Alivisatos and C. Larabell, Adv. Mater., 2002, 14, 882 CrossRef.
- Y. Wang, Z. Tang, S. Tan and N. A. Kotov, Nano Lett., 2005, 5, 243 CrossRef CAS.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225 CrossRef CAS.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11 CrossRef CAS.
- L. Cao, X. Wang, M. J. Meziani, F. S. Lu, H. F. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S. Y. Xie and Y. P. Sun, J. Am. Chem. Soc., 2007, 129, 11318 CrossRef CAS.
- (a) H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118 RSC; (b) S. -T. Yang, X. Wang, H. F. Wang, F. S. Lu, P. J. G. Luo, L. Cao, M. J. Meziani, J. -H. Liu, Y. F. Liu, M. Chen, Y. P. Huang and Y. -P. Sun, J. Phys. Chem. C, 2009, 113, 18110 CrossRef CAS; (c) F. Zhang, G. B. Braun, Y. F. Shi, Y. C. Zhang, X. H. Sun, N. O. Reich, D. Y. Zhao and G. Stucky, J. Am. Chem. Soc., 2010, 132, 2850 CrossRef CAS.
- (a) A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455 CrossRef CAS; (b) L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803 CrossRef CAS.
- J. Yang, F. Zhang, Y. Chen, S. Qian, P. Hu, W. Li, Y. Deng, Y. Fang, L. Han, M. Luqman and D. Zhao, Chem. Commun., 2011, 47, 11618 RSC.
- (a) K. Aslan, J. Huang, G. M. Wilson and C. D. Geddes, J. Am. Chem. Soc., 2006, 128, 4206 CrossRef CAS; (b) K. Aslan, P. Holley and C. D. Geddes, J. Mater. Chem., 2006, 16, 2846 RSC.
- C. D. Geddes and J. R. Lakowicz, J. Fluoresc., 2002, 12, 121 CrossRef.
- Y. Zhang, H. Goncalves, J. C. G. Esteves da Silva and C. D. Geddes, Chem. Commun., 2011, 47, 5313 RSC.
- (a) P. V. Kamat and B. Shanghavi, J. Phys. Chem. B, 1997, 101, 7675 CrossRef CAS; (b) J. Malicka, I. Gryczynski, J. Kusba and J. R. Lakowicz, Biopolymers, 2003, 70, 595 CrossRef CAS.
- J. Zong, Y. Zhu, X. Yang, J. Shen and C. Li, Chem. Commun., 2011, 47, 764 RSC.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- P. Y. Silvert, R. Herrera-Urbina, N. Duvauchelle, V. Vijayakrishnan and K. T. Elhsissen, J. Mater. Chem., 1996, 6, 573 RSC.
- C. Graf, D. L. J. Vossen, A. Imhof and A. V. Blaaderen, Langmuir, 2003, 19, 6693 CrossRef CAS.
- J. Zhang, Y. Fu, M. H. Chowdhury and J. R. Lakowicz, J. Phys. Chem. C, 2008, 112, 18 CAS.
- S. L. Westcott, S. J. Oldenburg, T. R. Lee and N. J. Halas, Langmuir, 1998, 14, 5396 CrossRef CAS.
- B. Zhang, C. Y. Liu and Y. A. Liu, Eur. J. Inorg. Chem., 2010, 4411 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |