Osteogenesis imperfecta mutations lead to local tropocollagen unfolding and disruption of H-bond network
Alfonso
Gautieri
ab,
Simone
Vesentini
b,
Alberto
Redaelli
b and
Markus J.
Buehler
*ac
aLaboratory for Atomistic and Molecular Mechanics (LAMM), Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Room 1-235A&B, Cambridge, MA, USA. E-mail: mbuehler@MIT.EDU; Tel: +1-617-452-2750
bBiomechanics Research Group, Department of Bioengineering, Politecnico di Milano, Via Golgi 39, 20133, Milan, Italy
cCenter for Computational Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA, USA
First published on 15th March 2012
Abstract
Osteogenesis imperfecta (OI), also known as “brittle bone disease”, is a rare genetic disorder of collagen tissues characterized by brittle bones and, in severe cases, prenatal death. Even though the macroscale consequences of the disease are well known, the effects of the mutations on the folding of collagen triple helix remain largely unknown. In this work we carry out metadynamics molecular simulations to estimate the effects of OI mutations on the folding of collagen triple helix, by quantifying the free energy changes as a function of the interchain distance, for all seven glycine substitutions known to lead to OI. We find that OI mutations lead to significant local unfolding in the vicinity of the mutation and that this phenomenon is more pronounced for mutations associated with the most severe OI phenotypes. We show that the mutation-induced unfolding leads to the disruption of the local interchain H-bonds, but the extent of the disruption is independent of the mutation type. We discuss our findings in the context of the three main OI mechanism models proposed in the literature, showing that the findings described here fit well in these models and, more importantly, could provide an important step towards a unified picture of OI aetiology.
Introduction
Collagen is a key construction material in vertebrate biology, formed through a hierarchical assembly of collagen molecules. Among the several forms of this protein, type I collagen is by far the most represented, being the major structural protein of connective tissues. Type I collagen molecule is a heterotrimer formed by two α1(I) and one α2(I) chains, encoded by the COL1A1 and COL1A2 genes, respectively. The chains associate in the rough endoplasmic reticulum using regions in the carboxyl-terminal propeptides and nucleate the triple helix, which is propagated linearly toward the amino terminal ends of the chains. The mature collagen molecule is transported to the Golgi, packaged into transport vesicles in which lateral aggregation, the initial phase of fibril formation, occurs, and the aggregates are then secreted into the pericellular space where mature collagen molecules self-assemble into fibrils with other matrix proteins.1Diseases associated with collagen abnormalities have severe consequences, because collagen is the structural protein within most human body tissues that are crucial for the mechanical integrity of organisms. Among the first reported mutations in collagen were those associated with the so-called “brittle bone disease” or osteogenesis imperfecta (OI), which involves a catastrophic breakdown of tissue.2 Patients affected by OI exhibit an array of symptoms, including short stature, loose joints, blue sclearae, dentinogenesis imperfecta, hearing loss, and neurological and pulmonary complications.3,4 The classification of OI is commonly based on clinical features described by Sillence et al. in 1979, which leads to four major groups, from mild (OI type I) to severe (OI type III and IV) to perinatal lethal (OI type II).5 The genetic basis for most forms of this disease lies in mutations of type I collagen genes, as tabulated in the database of human collagen mutations.6,7 Missense mutations that alter a glycine codon in COL1A1 or COL1A2 genes are the most common causes of OI.8 The replacement of either guanine (G) residue in the glycine codon GGC can theoretically result in the replacement of eight different amino acids: serine (Ser), cysteine (Cys), alanine (Ala), valine (Val), aspartic acid (Asp), glutamic acid (Glu), arginine (Arg) and tryptophan (Trp). All possibilities have been described in association with OI, although the frequency by which the different mutations occur varies considerably, with tryptophan replacements being exceedingly rare.9 Predicting the effect of specific mutations would help to guide genetic counseling decisions.10 However, even though some genotype–phenotype relationships are emerging,11–16 up until now the precise molecular mechanisms of how single point mutations can alter the structure of collagen molecules is missing. As a consequence, also how the molecular defect is propagated at larger tissue scales and how it affects the structure and properties of the tissue remain unknown, and in general the genotype–phenotype relationship remains evasive. There are some trends that are widely observed, such as the severity depending on the chain in which the Gly substitution occurs, the location within the chain, and the substituting amino acid,17 which have resulted in several different models for relating Gly substitutions to OI severity:
• Chain model. This early model18 focused on the chain in which the mutations occurred and postulated that phenotypes resulting from mutations on the α1(I) chain are more severe than those on the α2(I), because type I collagen heterotrimers contain two copies of the α1(I) chain, so that random assortment of mutant chains causes up to 75% of heterotrimers to contain one or more abnormal α1(I) chains vs. up to 50% of trimers with an abnormal α2(I) chain.
• Gradient model. This model emphasizes that changes within the C-terminal three-quarters of the molecule are usually more severe than those within the N-terminal region and implies a relation to the C-to-N directionality of collagen triple-helix folding. This model suggests that mutations in the triple helical domain delay the helix folding, resulting in additional hydroxylation and glycosylation of residues amino-terminal to the site of the substitution.17 However, while this model holds generally true for chain α1(I), it fails to explain the different severity outcome of mutations in the same position (but different type of substitution). Furthermore, it does not hold true for mutations affecting the α2(I) chain.
• Local model. To overcome limitations in the previous model, it has been suggested that the local environment of the amino acids at the mutation site is crucial in the severity of OI. Experimental and computational methods have been applied to predict lethality based on physicochemical properties of substituting residues and of the amino acids surrounding the mutation site.11,12,14,19–21 These investigations found that severity correlates with the identity of the substituting residue (where large and/or charged amino acids are more disruptive) and that local amino acid patterns partially correlate with lethality, where mutation of higher-stability triplets results in more severe disease. However, a statistically significant correlation between the stability of the mutated triplet and lethality was not found when this model was applied to an updated set of mutations.7
• Regional model. The identification of mutations not conforming to the previous patterns led to the proposal of the regional model, in which the location of the mutation, rather than the particular substituting residue, is decisive to the clinical outcome. In this model, the collagen D-period is divided into crucial and noncrucial regions interspersed along the collagen molecule. In particular, two region with almost exclusively lethal mutations (helix positions 691–823 and 910–964) align with major ligand-binding regions, suggesting that mutations could alter fundamental interactions of collagen monomers or fibrils with integrins, matrix metalloproteinases, fibronectin, and cartilage oligomeric matrix protein.7,22,23
The presence of several models of OI origin indicates that there is limited knowledge on the phenotype development and that the molecular mechanisms of how a single point mutation can alter the properties at single molecule level and consequently at larger tissue scales has yet to be elucidated. To address this point, we investigate the molecular scale effects of OI mutations on the structure and folding of the collagen molecule, then discussing how the structural changes at the molecular scale might affect the structure and properties of larger hierarchical structures.
We report a series of atomistic simulations to assess the effects of OI mutations on the folding of collagen triple helix. For this purpose, we consider an archetypical collagen peptide (i.e., with only Gly-Pro-Hyp triplets) as wild-type case and seven other cases, each with a different Gly substitution in the middle of one chain (see Fig. 1). We then perform metadynamics molecular simulations to monitor the free energy profile as a function of the local interchain distance (see Fig. 1B).
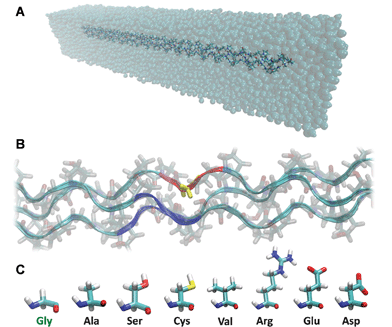 | ||
| Fig. 1 Atomistic model of the archetypical collagen peptides. Panel A shows the simulated system, with the collagen peptide in a water box. The central Gly residue of one chain (yellow in panel B) is mutated with the seven amino acids responsible for osteogenesis impefecta (panel C). During metadynamics simulations the free energy is calculated as a function of the distance between the chain bearing the mutation (i.e., the center of mass of the red region in panel B) and the center of mass of the other two chains (i.e., the center of mass of the blue regions in panel B). For the calculation of the interchain distance, we considered only backbone atoms of the residue bearing the mutation (or the corresponding on the non-mutated chains) and the immediate neighbor in both directions (thus a total of three amino acids per chain). | ||
The major goal addressed in this study is to elucidate and quantify the effect of OI mutations on the folding of mutant collagen peptides relative to the wild case and correlate the findings with the severity of OI, using archetypical collagen peptides. The hypothesis behind this work is that glycine substitutions lead to local unfolding of collagen molecules and that this defect relates to the identity of the mutation. This will help to advance the understanding of OI mechanism within the framework of the models discussed above, providing insights into the nanoscale defects on collagen folding caused by single point mutations.
Methods
Collagen model generation
The effect of osteogenesis imperfecta mutations on the tropocollagen folding is investigated using metadynamics simulations, using the well-tempered approach described in ref. 24. We use the THeBuScr (Triple-Helical collagen Building Script) code25,26 to build a model of the collagen molecule, as done in earlier studies.27–29 We choose the simplest model of collagen, with only Glycine-Proline-Hydroxyproline (GPO) triplets on each of the three chains in order to represent an archetype of collagen molecule, this triplet being the most represented along the triple helix (as well as the most stabilizing). The collagen model we use, [(GPO)21]3, is truncated to 63 amino acids per chain due to computational limitations, since the full-length collagen molecule (300 nm long) is too large for atomistic simulations. This leads to a collagen-like segment with a length of approximately 20 nm. Peptides of comparable length have been used both in earlier computational and experimental studies.13,19,28,30–33 In one of the three chains, the glycine residue in the central triplet (the 11th triplet) is mutated into one of the seven amino acids usually found as glycine substituents in osteogenesis imperfecta, namely alanine, serine, cysteine, valine, arginine, aspartic acid or glutamic acid. This leads to a total of eight cases (one wild case and seven mutated cases) (see Fig. 1B–C).Collagen model equilibration
Explicit water molecular dynamics simulations are performed using the NAMD code 2.734,35 patched with PLUMED36 and the CHARMM force field,37 including parameters for the non-standard hydroxyproline residue.38 Since the collagen triple helix considered here is truncated, the N-terminals are capped with ACE residues (acetylated N-termini), whereas C-terminals are capped with CT3 residues (amidated C-termini). The collagen peptides are solvated in a 22 nm × 5 nm × 5 nm water box using TIP3P water molecules for solvent, and ions are included to achieve a ionic concentration of 0.5 mol L−1 (see Fig. 1A). The total number of atoms of the solvated system is approximately 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms. Rigid bonds are applied to constrain covalent bond lengths, thus allowing a time step of 2 fs. Van der Waals interactions are computed using a cutoff for neighbor list at 1.35 nm, with a switching function between 1.0 and 1.2 nm. For electrostatic interactions a similar switching function is used for the dry case, whereas the Particle-Mesh Ewald sums (PME) method is applied to describe electrostatic interactions in the solvated systems. The preliminary energy minimization is performed by using a steepest descent algorithm until convergence. The systems are then equilibrated in NPT ensemble at a temperature of 310 K (37 °C) and at a pressure of 1 atm for 10 ns of molecular dynamics. We observe that the root-mean-square deviation (RMSD) of the protein backbone reaches a stable value within ≈5 ns simulation time, thus we assume that the collagen peptides are equilibrated properly at the end of the 10 ns molecular dynamics run. The final configurations of the proteins are used as starting points for the following metadynamics simulations.
000 atoms. Rigid bonds are applied to constrain covalent bond lengths, thus allowing a time step of 2 fs. Van der Waals interactions are computed using a cutoff for neighbor list at 1.35 nm, with a switching function between 1.0 and 1.2 nm. For electrostatic interactions a similar switching function is used for the dry case, whereas the Particle-Mesh Ewald sums (PME) method is applied to describe electrostatic interactions in the solvated systems. The preliminary energy minimization is performed by using a steepest descent algorithm until convergence. The systems are then equilibrated in NPT ensemble at a temperature of 310 K (37 °C) and at a pressure of 1 atm for 10 ns of molecular dynamics. We observe that the root-mean-square deviation (RMSD) of the protein backbone reaches a stable value within ≈5 ns simulation time, thus we assume that the collagen peptides are equilibrated properly at the end of the 10 ns molecular dynamics run. The final configurations of the proteins are used as starting points for the following metadynamics simulations.
Metadynamics simulations
Metadynamics simulations involve the definition of a set of collective variables (CVs) for which the associated free energy surface (FES) is calculated.39 Due to the presence of high barriers, it is necessary to use a sampling strategy – such as the introduction of a biasing potential – that forces the system to explore a region of high free energy. In metadynamics, the molecular dynamics run is biased by a history-dependent potential defined as the sum of Gaussian functions in the CV space.40 In the long time limit, the bias potential compensates the underlying free energy surface, providing an estimate of the free energy along the CVs. In well-tempered metadynamics, the height of the Gaussian potentials are automatically rescaled during the simulation, which guarantees that the bias potential converges in a single simulation and does not oscillate around the FES solution.24 In our study we monitor the free energy as a function of the distance between the chain bearing the mutation and the other two chains of the triple helix. Thus, we define the CV as the distance between the centre of mass of two groups of atoms. The first group is formed by the backbone atoms of the mutated amino acid and the two adjacent amino acids (for a total of 12 atoms). The second group is formed by the backbone atoms of the corresponding amino acids on the two chains that do not bear the mutation (thus, a total of 24 atoms) (see Fig. 1B). The Gaussian potentials are added every 1000 steps (corresponding to a time interval of 2 ps), and have an initial height of 0.1 kcal mol−1 and a width of 0.01 nm. The width of the Gaussian functions determines the resolution of the reconstructed free energy profile. Indeed, the sum of Gaussians efficiently reproduces features of the free energy profile on a scale larger than the width itself. In our case the resolution of the reconstructed free energy profile is therefore in the range of 0.1 nm.Metadynamics simulations are run in NPT ensemble at a temperature of 310 K (37 °C) and at a pressure of 1 atm for 100 ns, during which the biasing potential is collected. In order to monitor the convergence of the metadynamics runs, we plot the free energy profile every 4 ns and consider the calculation converged when the free energy profile does not change significantly as the simulation proceeds, i.e. we monitor how the free energy difference between the folded state (the free energy minimum) and an unfolded state changes in time. For the unfolded state we choose the point where the CV assumes a value of 1.1 nm, a condition of high free energy for all peptides. We assume to have reached convergence when this free energy difference is stable in time.
H-bond analysis
We monitor the number of H-bonds the chain bearing the mutation forms with the remaining two chains. In order to assess the interchain H-bonds we considered the triplet bearing the mutation plus the adjacent triplet in both directions and the corresponding triplets on the other two chains, thus a total of 9 amino acids per chain. We consider only the backbone donors and acceptors and determine H-bonds using a geometric definition, i.e. an angle donor-hydrogen-acceptor of 30° and a cutoff distance of 0.35 nm between the donor and the acceptor. Since there are three donors and three acceptors on the chain bearing the mutation, the total maximum number of H-bonds that this chain can form with the other two is six. In order to consider only relevant states, for the calculation of H-bonds we consider only the time frames in which the interchain distance is within ±0.1 nm of the energy minimum. H-bond analysis and in general visualization and imaging of molecular models has been performed using VMD 1.9.41Computational costs
Each of the eight solvated full-atomistic models contain ≈40000 atoms, and each metadynamics simulation has been run for 100 ns (time step 1 fs), requiring about 150 h on 32 CPUs on a parallel machine.
Results and discussion
The free energy profile for the reference case (no mutations, Fig. 2A) shows a distinct energy minimum for an interchain distance of 0.4 nm, in agreement with a molecular radius in the range 0.28–0.75 nm.42,43 Molecules with Gly mutations (Fig. 2B–H) display altered energy profiles, with a shallower and less defined energy minima, which suggest that the presence of a Gly substitution leads to a less stable configuration compared to the reference case. Furthermore, the energy minimum is shifted towards a larger distance, which implies a local unfolding (Fig. 3A–B). It is interesting to observe that in the case of Cys and Glu mutations there is a second energy minimum for a short interchain distance (≈0.2 nm). This corresponds to the configurations shown in Fig. 3C–D, where the side chain of the mutated amino acid points inwards with respect to the triple helix, laying between the other two chains. Although these configurations do not lead to an increased interchain distance, they lead to a local altered folding with respect to the healthy case, and this difference could be relevant for the disease mechanism. This is because the misfolding might interfere with the fibril formation, mineralization or docking of integrins or proteolgycans.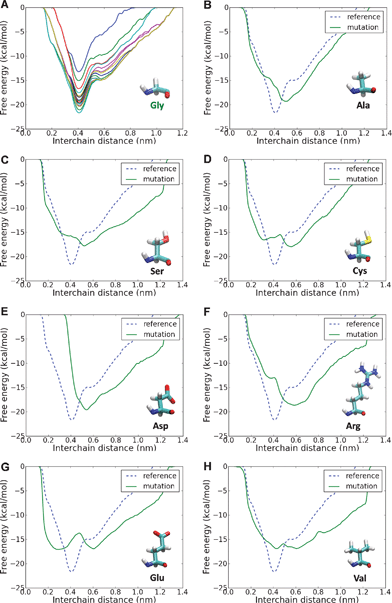 | ||
| Fig. 2 Free energy profile as a function of interchain distance for different glycine substitutions. In order to monitor the convergence of the metadynamics runs, we plot the free energy profile every 4 ns and consider the calculation converged when the free energy profile does not change significantly as the simulation proceeds. This is presented for the reference case (panel A), which shows that the free energy profile converges within the 100 ns simulations and presents a well-defined energy minimum for an interchain distance of 0.4 nm. The free energy profile is then calculated for the different OI-related mutations (panels B–F) and the profiles are compared to the reference case (dashed line). The results show that mutations hinder the well-defined energy minimum found in the reference cases, leading to a profile with a shallower minimum shifted to higher interchain distances, which reflects a destabilization of the local folding. | ||
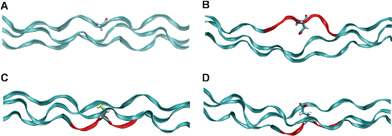 | ||
| Fig. 3 Energy minimum configurations. Energy minimum configuration is plotted for the reference case (panel A), where no mutation is introduced and the normal Gly residue (licorice rendering) leading to the triple helix folding of the collagen chains (ribbon rendering). When another amino acid (Asp, in panel B) substitutes the mandatory Gly residue, the energy minimum configuration shows a local unfolding of the triple helix, with an increasing distance between the chains. Regions marked in red are the section where there is a marked misfolding of the triple helix. The free energy profiles for both Cys and Glu mutations present two distinct energy minima: one for distances larger than the reference case (similar to the other mutations) and one, peculiar to these two cases, for distances shorter than the reference case. These metastable states correspond to a configuration in which the side chain of Cys (panel C) or Glu (panel D) points inwards with respect to the triple helix, laying between the other two chains. | ||
The main effect of mutation-induced misfolding is the disruption of the H-bonding between the collagen chains. In the vicinity of the mutation point, the chain bearing the mutation makes an average of 2.5 H-bonds with the other two chains in the reference case, whereas when a mutation is introduced the number of interchain H-bonds decrease sensibly, to an average ranging from 1.5 to 1.9 H-bonds, depending on the specific substitution. A deeper analysis of the interchain H-bonds shows that in the reference case the peptide spends the relative majority of the time forming three H-Bonds (out of six possible), with a small but significant time spent in configurations with a higher number of H-bonds. Conversely, when other amino acids replace the central Gly residue, typically forming 1 or 2 H-bonds, and the peptide rarely forms more than three H-bonds (see Fig. 4).
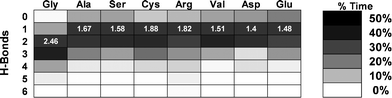 | ||
| Fig. 4 Effects of mutation type on interchain H-bonds. The most evident effect of the local unfolding is the disruption of the interchain H-bonding as shown by the average number of local backbone-backbone H-bonds, which decreases in the presence of a glycine substitution. The figure shows the percentage of time (depicted with gray scales) spent forming a specific number of H-bonds (where 6 is the maximum possible) for each of the 8 cases considered. The numbers in white give the average number of H-bonds formed. In the reference case the average is found to be 2.46 H-bonds, whereas in the mutated cases the average is shifted towards lower values. For the calculation of H-bonds we consider only the time frames in which the interchain distance is within ±0.1 nm of the energy minimum. | ||
Finally, we find that the changes at the molecular level, i.e. the local unfolding due to the presence of a Gly replacement, correlate strongly with the lethality associated to the specific mutations. Indeed, mutations that lead to a higher degree of unfolding (larger interchain distance) lead more often to lethal phenotype (OI type II). This is shown in Fig. 5 by a linear curve fit to the interchain distance at the energy minimum over the lethality of the specific mutation. The lethality is calculated as the ratio between the severe phenotype occurrences (leading to OI type II) and the total occurrences of each considered Gly replacement. An interesting case is the Gly-to-Val substitution. Indeed, the Val mutation, despite inducing a relatively moderate unfolding, it is associated with a high degree of lethal OI occurrences. In order to explain this phenomenon it is important to consider that the effects on the structure of the single collagen molecule are an important factor influencing the outcome of OI, but the single molecule level is not the only hierarchical scale affected by the mutations. Indeed, it is known that the presence of the mutations affect other phenomena such as intermolecular interaction, fibril formation and mineral crystal growth, which in turn determine the OI phenotype.7 Regarding the specific case of Val, in a previous work of our group focused on intermolecular interaction,11 we showed that Val represents the most disrupting case. These results suggest that Gly-to-Val mutation does affect the single molecule folding, but that the severity of the phenotype generally found in presence of this mutation is also due to higher scale effects. A further aspect to be considered is that the free energy minimum is very broad if compared to the Gly case, but also in comparison with the other mutations. This is another factor, along with the position of the minimum, which might explain the severity of Val mutations. Indeed, a deep minimum implies a stable configuration, i.e. a deviation from the energy minimum is unfavourable due to the energy barrier. Conversely, a broad minimum (such as the one found for Val) implies that the local configuration is unstable and that the chain separation can easily assume values higher than that associated with the energy minimum. Thus, an energy minimum shifted toward higher interchain distances together with an unstable configuration might be concurrent factors leading to the high severity observed for Val mutations.
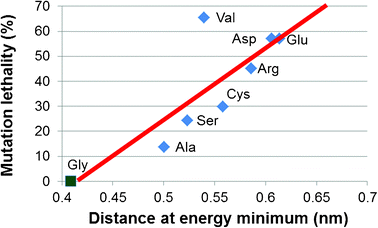 | ||
| Fig. 5 OI phenotype correlates with collagen unfolding. Glycine substitutions lead to local unfolding of the collagen triple helix, measured by the increase in the local interchain distance, which correlates well with the lethality of the mutations. The lethality is given by the percentage of OI type II (the most severe/lethal form) cases for each type of mutation. Data on lethality are taken from ref. 7. | ||
The main result of our work is that it showed that OI mutations lead to local unfolding of the triple helix, which is assessed through the free energy profile as a function of the interchain distance. We also show that this local unfolding has a strong correlation with the severity of the phenotype associated with the specific mutation. In the past, both experimental13 and computational44 works have used collagen peptides similar to the one used in this work to assess the effects of point mutations on single collagen molecule. In these works, the authors obtained the melting free energy (the difference in free energy between the triple helix and the separate single chains) and used this quantity as a measure of the stability of collagen peptides, showing that the reference peptide (without mutation) is the most stable. When mutations are introduced, it is observed that there is a strong correlation between the decrease in the melting free energy and the severity of the mutation, i.e. mutations associated with higher severity lead to lower melting temperatures (which means that the peptide is less stable). These results match our findings rather well, as depicted in Fig. 5, providing support to our conclusions. A possible explanation is that the mutation-induced local unfolding we observe in the collagen peptides is the responsible for the decreased stability with respect to melting observed in previous studies. Indeed, the disruption of increased interchain distance, with the corresponding hindrance of interchain H-bonding could likely lead to an easier melting of the triple helix.
In our own previous works12,45 we found that OI mutations lead to a decrease of the tensile modulus (up to 15%) of the collagen peptides. The reason for this softening may be explained considering the local unfolding and the consequent partial disruption of backbone-backbone H-bonds between the chains, which would act as a local defect and hinder the mechanical resistance of the molecule.
Our results, obtained here using a more advanced sampling method, strengthen the local model for OI mechanism, showing that the disruption of the helical folding is well correlated with the identity and biophysical properties of the substituting amino acids. Indeed, we show that small Gly replacements (Ala, Ser) lead to a less extended unfolding (with an interchain distance at the energy minimum of ≈0.5 nm) when compared to charged or large Gly replacements (Arg, Asp or Glu), which lead to more pronounced unfolding (interchain distance at the energy minimum of ≈0.6 nm). It is not clear how the local unfolding found here could lead to the symptoms of OI, such as bone brittleness and tendon laxity. The local unfolding may play a role in the collagen supramolecular assembly, disturbing the packing of collagen fibrils and leading to a partial loss of the lateral crystallinity, a feature often observed in OI tissues.46,47 In turn, a poorer lateral packing of collagen may affect the growth of mineral crystals, which may fail to grow highly organized in the fibril axis direction. Rather, they might be less organized and round-shaped instead of platelet-like. This feature would be consistent with the observed size and shape of the mineral crystals found in OI.48–50
The molecular effect of OI mutations described in this work can be of help to support and explain other OI models. In the framework of the gradient model, a mutation close to the C-terminus is shown to be highly disruptive, irrespective of the type of mutation.14 As we shown here, the presence of a mutation hinders the formation of interchain H-bonds, which are crucial for the stability of the triple helical folding. Thus, if the mutation falls in the C-terminal region, where the zip-like folding onsets, the lack of H-bonds may block or severely delay the collagen folding process. Conversely, if the mutation affects another region, with the collagen already partially folded, the effect might be minor. In this respect, it is interesting to observe that the degree of unfolding has a correlation with the identity of the mutation, whereas the disruption of H-bonds does not depend on the mutation. Indeed, our results show that the H-bond disruption follows an on/off mechanism, and all types of mutations significantly decrease the ability of the peptide to form local interchain H-bonds. This could explain why in the gradient model the type of mutation has none or marginal importance.
Finally, there are two regions along the collagen molecule (helical position 691–823 and 910–964) in which essentially only lethal substitutions are identified. These long stretches of lethal mutations correlate strongly with two (out of three) collagen major ligand binding regions,7,51 suggesting a regional model for OI. Molecular recognition necessitates that the interacting molecules are folded properly, so that the binding sites can connect. Thus, the mutation-induced unfolding, when located in a ligand binding region might alter the binding site configuration, impeding the correct docking of extracellular molecules (such as proteoglycans or integrins) to collagen.
Conclusions
Since the identification of OI by Sillence et al. in 1979 there has been a remarkable effort in the investigation of OI and its origin. Several details of the effects of OI at various scales are now known and a number of models have been proposed to depict the molecular mechanism responsible for the disease onset. However, despite great efforts, a unified model of the OI mechanisms is still lacking and even the effects of OI on collagen’s molecular structure are unclear.The main focus of this work was to assess the quantitative effect of OI-related glycine replacement mutations on the molecular folding of collagen molecule. We found that OI mutations have a strong effect on collagen at the molecular hierarchy and we discuss these findings in the contest of the previously proposed model for OI mechanisms, showing that our results can provide a basis to explain and unify the three major models. We showed that the local unfolding is strongly dependent on the substituting amino acids, providing further basis for the so-called local model. On the other hand, we also demonstrated that the rupture of H-bonds (which are essential for the correct zip-like folding of the three chains in collagen molecules) is insensitive to the type of mutation, providing ground to explain the gradient model, in which the position to the C-terminus is the key issue, whereas the type of mutation is less important. Finally, since the correct folding is of paramount importance for molecular recognition, we suggest that local unfolding caused by mutations could severely affect the binding of extracellular molecules, as implied in the regional model.
The present study resulted in a detailed and quantitative insight into the effect of OI-related mutations at the molecular scale, and clearly showed that severity of the OI mutation types and the degree of unfolding are strongly correlated. Our findings also provide likely explanations of the three major OI models proposed in the literature and could thus be critical on the path of forming a more holistic picture of OI aetiology.
Acknowledgements
We acknowledge support from the MIT-Italy Program (“Progetto Rocca”), NSF-CAREER (award number 0642545), DOD-PECASE (award number N000141010562) and Politecnico di Milano (Grant “5 per mille junior 2009”). High-performance computing resources have been provided by Regione Lombardia and CILEA Consortium through the LISA Initiative and by CINECA under the ISCRA initiative.References
- K. E. Kadler, C. Baldock, J. Bella and R. P. Boot-Handford, J. Cell Sci., 2007, 120, 1955–1958 CrossRef CAS.
- L. Peltonen, A. Palotie and D. J. Prockop, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6179–6183 CrossRef CAS.
- F. Rauch and F. H. Glorieux, Lancet, 2004, 363, 1377–1385 CrossRef CAS.
- D. Primorac, D. W. Rowe, M. Mottes, I. Barisic, D. Anticevic, S. Mirandola, M. G. Lira, I. Kalajzic, V. Kusec and F. H. Glorieux, Croat. Med. J., 2001, 42, 393–415 CAS.
- D. O. Sillence, A. Senn and D. M. Danks, J. Med. Genet., 1979, 16, 101–116 CrossRef CAS.
- R. Dalgleish, Nucleic Acids Res., 1997, 25, 181–187 CrossRef CAS.
- J. C. Marini, A. Forlino, W. A. Cabral, A. M. Barnes, J. D. San Antonio, S. Milgrom, J. C. Hyland, J. Korkko, D. J. Prockop, A. De Paepe, P. Coucke, S. Symoens, F. H. Glorieux, P. J. Roughley, A. M. Lund, K. Kuurila-Svahn, H. Hartikka, D. H. Cohn, D. Krakow, M. Mottes, U. Schwarze, D. Chen, K. Yang, C. Kuslich, J. Troendle, R. Dalgleish and P. H. Byers, Hum. Mutat., 2007, 28, 209–221 CrossRef CAS.
- K. M. Kozloff, A. Carden, C. Bergwitz, A. Forlino, T. E. Uveges, M. D. Morris, J. C. Marini and S. A. Goldstein, J. Bone Miner. Res., 2004, 19, 614–622 CrossRef.
- P. J. Roughley, F. Rauch and F. H. Glorieux, Eur. Cells Mater., 2003, 5, 41–47 CAS.
- W. A. Cabral, S. Milgrom, A. D. Letocha, E. Moriarty and J. C. Marini, J. Med. Genet., 2006, 43, 685–690 CrossRef CAS.
- A. Gautieri, S. Uzel, S. Vesentini, A. Redaelli and M. J. Buehler, Biophys. J., 2009, 97, 857–865 CrossRef CAS.
- A. Gautieri, S. Vesentini, A. Redaelli and M. J. Buehler, Protein Sci., 2008, 18, 161–168 Search PubMed.
- K. Beck, V. C. Chan, N. Shenoy, A. Kirkpatrick, J. A. M. Ramshaw and B. Brodsky, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4273–4278 CrossRef CAS.
- D. L. Bodian, B. Madhan, B. Brodsky and T. E. Klein, Biochemistry, 2008, 47, 5424–5432 CrossRef CAS.
- B. Brodsky, M. A. Bryan and H. M. Cheng, Biopolymers, 2011, 96, 4–13 CrossRef.
- E. Makareeva, W. A. Cabral, J. C. Marini and S. Leikin, J. Biol. Chem., 2006, 281, 6463–6470 CrossRef CAS.
- P. H. Byers, Philos. Trans. R. Soc. London, Ser. B, 2001, 356, 151–157 CrossRef CAS.
- D. J. Prockop, C. D. Constantinou, K. E. Dombrowski, Y. Hojima, K. E. Kadler, H. Kuivaniemi, G. Tromp and B. E. Vogel, Am. J. Med. Genet., 1989, 34, 60–67 CrossRef CAS.
- A. Persikov, J. Ramshaw, A. Kirkpatrick and B. Brodsky, Biochemistry, 2000, 39, 14960–14967 CrossRef CAS.
- W. Yang, M. L. Battineni and B. Brodsky, Biochemistry, 1997, 36, 6930–6935 CrossRef CAS.
- K. H. Lee, K. Kuczera and M. M. Holl, Biophys. Chem., 2011, 156, 146–152 CrossRef CAS.
- J. C. Marini, M. B. Lewis, Q. Wang, K. J. Chen and B. M. Orrison, J. Biol. Chem., 1993, 268, 2667–2673 CAS.
- Q. Wang, B. M. Orrison and J. C. Marini, J. Biol. Chem., 1993, 268, 25162–25167 CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, DOI:10.1103/PhysRevLett.100.020603.
- J. Rainey and M. Goh, Protein Sci., 2004, 13, 2276–2276 CrossRef CAS.
- J. Rainey and M. Goh, Bioinformatics, 2004, 20, 2458–2459 CrossRef CAS.
- M. Srinivasan, S. G. M. Uzel, A. Gautieri, S. Keten and M. J. Buehler, Math. Mech. Solids, 2010, 15, 755–770 CrossRef.
- A. Gautieri, S. Vesentini, A. Redaelli and M. J. Buehler, Int. J. Mater. Res., 2009, 100, 921–925 CrossRef CAS.
- A. Gautieri, A. Russo, S. Vesentini, A. Redaelli and M. J. Buehler, J. Chem. Theory Comput., 2010, 6, 1210–1218 CrossRef CAS.
- A. V. Persikov, J. A. Ramshaw and B. Brodsky, Biopolymers, 2000, 55, 436–450 CrossRef CAS.
- M. Srinivasan, S. G. M. Uzel, A. Gautieri, S. Keten and M. J. Buehler, J. Struct. Biol., 2009, 168, 503–510 CrossRef CAS.
- A. Gautieri, S. Vesentini, F. M. Montevecchi and A. Redaelli, J. Biomech., 2008, 41, 3073–3077 CrossRef.
- P. J. Veld and M. J. Stevens, Biophys. J., 2008, 95, 33–39 CrossRef.
- M. T. Nelson, W. Humphrey, A. Gursoy, A. Dalke, L. V. Kale, R. D. Skeel and K. Schulten, Int. J. High Perform. Comput. Appl., 1996, 10, 251–268 CrossRef.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- S. Park, R. J. Radmer, T. E. Klein and V. S. Pande, J. Comput. Chem., 2005, 26, 1612–1616 CrossRef CAS.
- M. Parrinello and A. Laio, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef.
- T. Huber, A. E. Torda and W. F. van Gunsteren, J. Comput.-Aided Mol. Des., 1994, 8, 695–708 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- K. Beck and B. Brodsky, J. Struct. Biol., 1998, 122, 17–29 CrossRef CAS.
- P. Fratzl, N. Fratzlzelman and K. Klaushofer, Biophys. J., 1993, 64, 260–266 CrossRef CAS.
- K. H. Lee and M. M. Holl, Biopolymers, 2011, 95, 401–409 CrossRef CAS.
- A. Gautieri, S. Vesentini, F. M. Montevecchi and A. Redaelli, J. Biomech., 2008, 41, 3073–3077 CrossRef.
- D. J. McBride, V. Choe, J. R. Shapiro and B. Brodsky, J. Mol. Biol., 1997, 270, 275–284 CrossRef CAS.
- A. Miller, D. Delos, T. Baldini, T. M. Wright and N. P. Camacho, Calcif. Tissue Int., 2007, 81, 206–214 CrossRef.
- B. Grabner, W. J. Landis, P. Roschger, S. Rinnerthaler, H. Peterlik, K. Klaushofer and P. Fratzl, Bone, 2001, 29, 453–457 CrossRef CAS.
- N. P. Camacho, L. Hou, T. R. Toledano, W. A. Ilg, C. F. Brayton, C. L. Raggio, L. Root and A. L. Boskey, J. Bone Miner. Res., 1999, 14, 264–272 CrossRef CAS.
- P. Fratzl, O. Paris, K. Klaushofer and W. J. Landis, J. Clin. Invest., 1996, 97, 396–402 CrossRef CAS.
- S. M. Sweeney, J. P. Orgel, A. Fertala, J. D. McAuliffe, K. R. Turner, G. A. Di Lullo, S. Chen, O. Antipova, S. Perumal, L. Ala-Kokko, A. Forlino, W. A. Cabral, A. M. Barnes, J. C. Marini and J. D. San Antonio, J. Biol. Chem., 2008, 283, 21187–21197 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |