Coordination chemistry of titanium and zinc in Ti(1−x)Zn2xO2 (0 ≤ x ≤ 1) ultrathin films grown by DC reactive magnetron sputtering
Ignacio
Caretti
*,
Miriam
Yuste
,
Ricardo
Torres
,
Olga
Sánchez
,
Ignacio
Jiménez
and
Ramón
Escobar Galindo
Instituto de Ciencia de Materiales de Madrid (ICMM-CSIC), Campus de Cantoblanco, 28049, Madrid, Spain. E-mail: caretti@icmm.csic.es; Fax: +34 91 372 0623; Tel: +34 91 334 9000
First published on 17th February 2012
Abstract
XANES and RBS were used to explore the structural phase and composition transitions of ultrathin films with Ti(1−x)Zn2xO2 stoichiometries, from very low (∼0.5 at.%) to high (∼25 at.%) titanium content. In this way, the coordination chemistry of Ti4+ and Zn2+ cations was examined for a wide range of oxide compositions.
Transition metal doping and the formation of mixed oxides are widely used mechanisms to improve the intrinsic properties of binary oxides. Both procedures have been decisive in explaining the spectacular increase of applications based on ZnO and TiO2 films.
Proven applications of metal-doped ZnO in optoelectronic devices include photovoltaic and dye sensitized solar cells, flat panel displays, photodetectors, gas sensors, light emitting diodes and blue laser diodes.1–4 The vast majority of previous work on doped ZnO films is related to doping with group III elements. In particular Al, Ga and In trivalent cations have been extensively used to enhance the n-type conductivity of ZnO films.5–11 Fewer studies have been performed on quadrivalent dopants, such as titanium, which have one more valence than the trivalent cations and can provide two free electrons per atom to improve the conductivity of the ZnO host. In particular the Ti4+ cation has a slightly smaller radius (6.8 nm) than Zn2+ (7.4 nm) and, thus, it can act as a suitable donor in ZnO films. Some authors have reported an enhancement of the conductivity in Ti–ZnO (TZO) films for low titanium content (<1.5 at.%).12–16 However, the conduction mechanism of the metal–semiconductor transition in Ti-doped ZnO is still unclear.17–18
In turn, ZnO–TiO2 mixed oxides have been widely studied for use as gas sensors,19–20 antireflective coatings in solar cells,21–22 in photocatalytic and self-cleaning applications showing an improved photocatalytic activity and an extended absorption threshold towards the visible range23 and, more recently, as promising candidates for high frequency dielectric materials24,25 or novel luminescent phosphors.26 The mechanisms driving the formation of three different zinc titanate compounds in the ZnO–TiO2 system, namely, zinc ortho-titanate Zn2TiO4 (cubic inverse spinel crystal structure with a unit cell a = 0.846 nm), zinc meta-titanate ZnTiO3 (either cubic inverse defect spinel, a = 0.840 nm or hexagonal ilmenite structure, a = 0.507 nm) and Zn2Ti3O8 (cubic inverse defect spinel, a = 0.843 nm) are still under debate.27–29
Despite the extensive studies performed on the individual TZO and ZnO–TiO2 systems in areas such as photovoltaics, photocatalysis or optoelectronics, there is a lack of systematic work to fully understand Ti incorporation into the ZnO structure either as a dopant or to form a mixed oxide. In particular, one of the current technological challenges is the elucidation of such mechanism for nanostructured and ultrathin films due to the size-dependent electronic and optical properties of these semiconductor materials, as discussed in ref. 30. The scarce structural studies performed on these systems by XANES have used high energy photons to excite the K-edges of Ti and Zn.17,31 However, it is very informative to study the L-edges because their features are sharper and better resolved. In this work we have performed XANES at the Ti–L, Zn–L and O–K edges to explore the transition among those regimes (doped versus mixed oxide) by increasing the Ti load in ZnO films with thicknesses in the 10–40 nm range.
Ti(1−x)Zn2xO2 (0 ≤ x ≤ 1) samples were prepared by DC reactive magnetron sputtering on high resistivity (>6000 Ω cm) silicon wafers with a (100) orientation. Even though no intentional heating was applied to the substrate, a temperature between 90–100 °C was reached during deposition due to the impinging sputtered atoms, as measured with a thermocouple in contact with the sample. The samples were grown introducing a mixture of Ar (99.999%) and O2 (99.992%) gases in the vacuum chamber using a relative oxygen gas flow of 20% and a total flow (Ar + O2) of 30 sccm (standard cubic centimetres per minute). All the samples were prepared after complete poisoning of both cathodes. Firstly, different samples were deposited maintaining the Zn cathode power (PZn) at 100 W and increasing the Ti cathode power (PTi) from 0 to 120 W. Secondly, PTi was maintained at 100 W increasing PZn from 0 to 100 W. In this way, ultrathin films with thicknesses between 13 and 38 nm were obtained using a constant deposition time of 6 min. Further details on the experimental setup and procedure can be found elsewhere.32
For the study of the chemical composition of the films, Rutherford backscattering spectroscopy (RBS) experiments were carried out with the 5 MV HVEE Tandetron accelerator sited at the Centro de Micro-Análisis de Materiales (CMAM) of Universidad Autónoma de Madrid.33 The RBS experiments were performed using 3.035 MeV He+ ions to make use of the cross section resonance 16O (α, α) 16O at that particular energy and, therefore, to improve the sensitivity to oxygen. The chemical composition values have been extracted using the RBX software.34 The X-ray absorption near edge structure (XANES) measurements were done at the PM4 beamline of the Bessy II synchrotron facility (Berlin, Germany), using the SurICat endstation. The XANES spectra were recorded in the total electron yield (TEY) mode by registering the current drained to ground from the sample. The latter was normalized to the current from a gold-coated grid located upstream in the X-ray beam path.
Fig. 1 shows the XANES survey and RBS spectra of two selected samples with low (1 at.%) and high (25 at.%) Ti content, which were deposited using relative target powers of PZn/PTi = 100 W/40 W and 10 W/100 W, respectively (see Table 1). Pure ZnO and TiO2 reference spectra are also plotted in Fig. 1B for comparison purposes. In Fig. 1A the Ti L and O K-edges are presented, together with zinc's second order signal. Fig. 1B displays the RBS spectra (experimental data and the global fit results) with a zoom out (×20) of the area where the metal elements are observed (E = 2000–2500 keV). Both techniques are suitable to detect low metal contents in the deposited ultrathin films. The quantification of the elemental composition of all samples by XANES and RBS is summarized in Table 1, where the relative target power is also indicated. In general, there is a good agreement between the quantification by both techniques, taking into account that RBS quantifies the whole film, whereas XANES only maps the outermost ∼5 nm region.
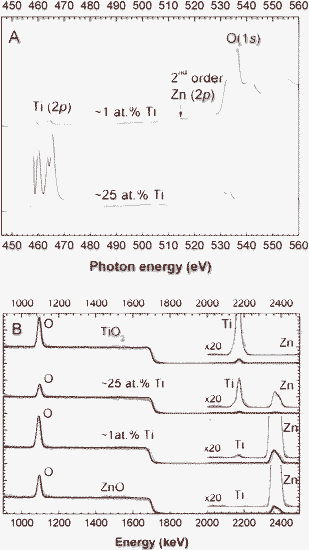 | ||
| Fig. 1 XANES survey (A) and RBS spectra (B) of samples with ∼1 at.% and ∼25 at.% of Ti. Reference RBS spectra for TiO2 and ZnO materials are also included. | ||
| Cathode power PZn/PTi (W/W) | RBS (at.%) | XANES (at.%) | ||||
|---|---|---|---|---|---|---|
| Ti | Zn | O | Ti | Zn | O | |
| 100/10 | 0.3 | 45.5 | 54 | 0.5 | 45.9 | 53.6 |
| 100/40 | 0.9 | 44 | 55.1 | 1.3 | 40.0 | 58.7 |
| 100/60 | 1.3 | 45.5 | 53.2 | 0.7 | 35.3 | 64.0 |
| 100/80 | 1.5 | 44 | 54.5 | 2.4 | 36.1 | 61.5 |
| 100/100 | 1.9 | 43.6 | 54.5 | 3.6 | 38.2 | 58.2 |
| 100/120 | 2.4 | 43.5 | 54.1 | 3.1 | 36.0 | 60.9 |
| 10/100 | 27.3 | 16.4 | 56.3 | 22.0 | 27.1 | 50.9 |
| 40/100 | 6.5 | 37.8 | 55.7 | 6.6 | 41.4 | 52.0 |
| 60/100 | 3.7 | 50.6 | 45.7 | 3.2 | 34.0 | 62.8 |
| 80/100 | 1.5 | 44.4 | 54.1 | 2.8 | 39.6 | 57.6 |
| 100/100 | 1.8 | 43.6 | 54.6 | 3.6 | 38.2 | 58.2 |
Fig. 2 shows the variation of the titanium atomic content (%) in the films obtained by XANES (open circles) and RBS (close circles). For the sake of simplicity, we will use the composition as measured by RBS in the discussion of Fig. 2. For the samples deposited with a fixed PTi = 100 W and PZn < 60 W, there is an abrupt decrease of the titanium content from ∼30 ± 1 at.% down to ∼4.0 ± 0.5 at.%. For higher Zn cathode powers the amount of titanium remains fairly constant around 2.0 at.%. In the cases where the samples are deposited with a constant Zn cathode power PZn = 100 W, the titanium content slightly and linearly increases with the Ti cathode power from 0 to 2.4 at.%.
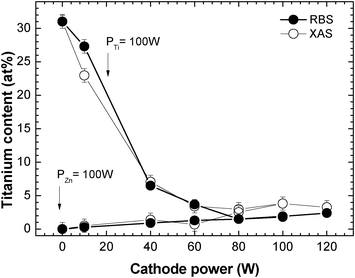 | ||
| Fig. 2 Variation of the titanium atomic content (at.%) in the Ti–Zn–O thin films with the Zn and Ti cathode powers for a fixed Ti and Zn powers of 100 W, respectively. | ||
As a whole, regardless of the cathode power, the Ti concentration in the films always stays below 7 at.%, except for the one deposited with PZn/PTi = 10 W/100 W. The incorporation of Ti in the films is controlled by the very low sputtering rate of Ti compared to Zn. Hence, we observe a significant increase in Ti concentration only for low Zn cathode powers below 40 W. Next, we will elucidate how these changes observed in the composition of the samples are reflected in the bonding structure of the formed compounds.
Fig. 3 shows the XANES spectra of selected Ti(1−x)Zn2xO2 samples for decreasing Ti concentration, from 22 at.% to 0.5 at.%, together with the reference spectra of anatase TiO2 and wurtzite w-ZnO thin films grown with the same sputtering setup. The composition values indicated correspond to the XANES data of Table 1. The Ti L-edge XANES spectrum of barium orthotitanate (Ba2TiO4) and the Zn L-edge XANES spectrum of zinc sulphate (ZnSO4) have been taken from the literature for comparison purposes.35,36
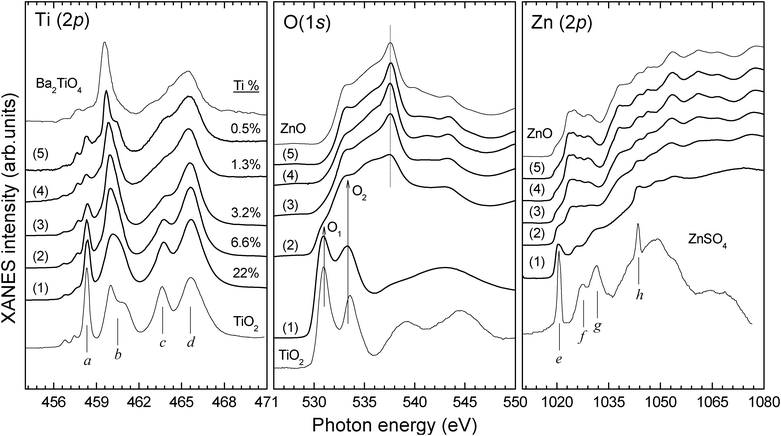 | ||
| Fig. 3 Ti L-, Zn L- and O K-edge XANES spectra of TZO ultrathin films. Titatium at.% concentrations are indicated as calculated by XANES (see Table 1). Anatase TiO2 and wurtzite ZnO thin films grown with the same sputtering setup are included, together with the Ti L-edge XANES spectrum of barium orthotitanate (Ba2TiO4)35 and the Zn L-edge XANES spectrum of zinc sulphate (ZnSO4).36 | ||
In the case of anatase TiO2, the Ti L-edge peaks (a,b) and (c,d) correspond to the (t2g, eg) sublevels in the L3- and L2-edges, respectively, where the characteristic peak b splitting arises from strong distortion of the Ti4+ octahedral symmetry.37 Spectrum (1) shows that, for a Ti content of 22 at.% in the films, the Ti L-edge is similar to that of pure anatase TiO2. However, the considerably smaller b splitting in sample (1) indicates a higher octahedral symmetry compared to anatase, which is typical of titanate phases.37 Moreover, the Zn L-edge of spectrum (1) exhibits 4 features distinctive of a zinc sulphate like structure (isostructural to Cu2SO4), which are labelled e–h in the reference spectrum. Zn2+ cations are thus coordinated to oxygen in octahedral symmetry, with a coordination number of 6. These observations suggest that sample (1) has a hexagonal h-ZnTiO3 ilmenite type structure formed by ZnO6 and TiO6 octahedrons. Zn L- and O K-edges of zinc spinel compounds38,39 are completely different to those in spectrum (1), excluding the formation of other known zinc titanate phases with inverse spinel structure (e.g. cubic ZnTiO3 or Zn2TiO4). Furthermore, the O K-edge line shape is consistent with an ilmenite-like structure.40 Remarkably, even at this high Zn concentration (27 at.%), the O K-edge XANES contains no w-ZnO signal. A decrease in the separation of peaks O1 and O2 compared to anatase TiO2 indicates a lower crystal field splitting value, as expected for the mentioned reduction of the octahedral distortion in titanates. Curiously, due to their structural similarities, an h-ZnTiO3 like phase should generally be obtained from a rutile TiO2 precursor and not anatase (the phase obtained here without Zn sputtering).41
For a considerably lower Ti content of 6.6 at.% (spectrum (2)), the Zn L-edge XANES spectrum is now quite similar to w-ZnO, but still exhibits weak hints of the e–h features from ZnO6 octahedrons. Besides, the significant narrowing and a downwards energy shift of peak b in the Ti L-edge of spectrum (2) points to a further reduction of the octahedral distortion and of the resulting crystal field splitting (related to the a–b peak separation).37 Again, these changes are characteristic of the Ti L-edge XANES spectrum of a titanate compound containing TiO6 units. Regarding the O K-edge, clear contributions from both w-ZnO and TiO6 environments are observed, what indicates the formation of a mixed oxide structure. Hence, we assign spectrum (2) to the formation of a w-ZnO/h-ZnTiO3 mixed-phase oxide.
Further reduction of the Ti concentration produces a significant intensity reduction of peaks a and c in the Ti L-edge XANES of samples (3) to (5). Additionally, peak b keeps narrowing and shifting to lower photon energies. These changes are explained by a gradual modification of the Ti site geometry from octahedral to tetrahedral symmetry and the consequent reduction in the coordination number from 6 to 4. The same transformation has been reported for the Ti L-edge XANES spectra of BaTiO3 (octahedral symmetry) and Ba2TiO4 (tetrahedral symmetry).35 At this point, we cannot entirely exclude the formation of a 5-coordinate square pyramidal Ti species (e.g. a fresnoite structure42) in this transition from high to low Ti content in the TZO films. The Zn L- and O K-edges of spectra (3)–(5) are almost identical and characteristic of w-ZnO (see reference spectrum). Moreover, as in pure ZnO, samples (3)–(5) reveal a dominant O peak at ∼537.5 eV from transitions to O 2pz and O 2px![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +y states,43 which is indicative of O-terminated surfaces.44 Thus, the XANES results indicate that for Ti concentrations below 3 at.% the Ti atoms incorporate in the ZnO structure with tetrahedral symmetry by substituting the Zn2+ cations in the oxide lattice, that is, forming a Ti-doped w-ZnO phase. Interestingly, we observe again a splitting of peak b for the lowest Ti concentration (0.5 at.%) that may be related with a distortion of the tetrahedral Ti4+ coordination symmetry due to differences in the Ti–O and Zn–O bond distances.
+y states,43 which is indicative of O-terminated surfaces.44 Thus, the XANES results indicate that for Ti concentrations below 3 at.% the Ti atoms incorporate in the ZnO structure with tetrahedral symmetry by substituting the Zn2+ cations in the oxide lattice, that is, forming a Ti-doped w-ZnO phase. Interestingly, we observe again a splitting of peak b for the lowest Ti concentration (0.5 at.%) that may be related with a distortion of the tetrahedral Ti4+ coordination symmetry due to differences in the Ti–O and Zn–O bond distances.
A preliminary study of the electrical properties of the TZO films was carried out with the four-point probe method. As reference value, the undoped ZnO sample has a resistivity of 1.52 Ω cm. Interestingly, the TZO coating with a 0.5 at.% Ti concentration gave the lowest resistivity of 4.5 × 10−2 Ω cm for our samples. Thus, the optimum behaviour of Ti as an electrical dopant takes place for concentrations below 1 at.%. A similar behaviour, but with a Ti load threshold of 1.3 at.%, has been reported for 500 nm16 and 650 nm17 thick TZO films, with optimum resistivities of 9.02 × 10−3 Ω cm and 3.82 × 10−3 Ω cm, respectively. TZO resistivity values found in the literature13–17 are generally smaller than the ones reported in this communication for ultrathin TZO films, whose significantly lower thickness may result in increased resistance values due to larger electron scattering effects. In fact, an increase of one order of magnitude in the resistivity has been described for decreasing thicknesses of TZO coatings from 950 nm to 30 nm.14 Our resistivity measurements are comparable to the 30 nm thick TZO in ref. 14 and to recently reported Ti doped ZnO nanowires.45 At this stage, we have not identified the type and concentration of defects present in our samples. Therefore, we have decided to avoid any speculative arguments concerning the defect chemistry and its connection to the conduction mechanisms in TZO.
According to the above, ZnO samples doped with a 0.5 at.% of Ti and thicknesses down to 10 nm may be suitable for electronic and optoelectronic devices, solar cells, photoelectrodes, photocatalysts or sensors.
Conclusions
Ultrathin films with Ti(1−x)Zn2xO2 (0 < x ≤ 1) compositions were deposited at room temperature by DC reactive magnetron sputtering on (100) Si wafers. We observe a transition from octahedrally coordinated Zn2+ in a h-ZnTiO3 like structure to the expected tetrahedral symmetry in the w-ZnO matrix of TZO films. Regarding Ti4+, a similar change from octahedral to tetrahedral sites is detected in the transition from h-ZnTiO3 to TZO coatings. Therefore, Ti is incorporated as a dopant by substituting Zn atoms in the wurtzite ZnO structure. The optimum Ti doping was found to be 0.5 at.%, for which the TZO sample showed a much lower resistivity than the more insulating ZnO films.Acknowledgements
We are indebted to A. Vollmer for her help at the Optics beamline (SURICAT endstation) of the Helmholtz-Zentrum Berlin-Electron storage ring BESSY II. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement n.226716. This work has been partially financed by the Spanish MICINN through project Consolider FUNCOAT CSD2008-0023, and project MOCASIN MAT2010-21070-C02-02. I. Caretti acknowledges support from CSIC through a JAE-Doc fellowship. R. Escobar Galindo would like to thank the MCINN for the financial support within the Ramón y Cajal programme (RyC2007-0026).References
- K. Ellmer and A. Klein in Transparent Conductive Zinc Oxide: Basics and Applications in Thin Film Solar Cells, ed. K. Ellmer, A. Klein, B. Rech, Springer, New York, 2008, ch. 1, 1–27 Search PubMed.
- T. Minami, MRS. Bull., 2000, 25, 38 CrossRef CAS.
- T. Kamiya and M. Kawasaki, MRS Bull., 2008, 33, 1061 CrossRef CAS.
- H. Ohta and H. Hosono, Mater. Today, 2004, 7, 42 CrossRef CAS.
- A. Anders, S. H. N. Lim, K. M. Yu, J. Andersson, J. Rosén, M. McFarland and J. Brown, Thin Solid Films, 2010, 518, 3313 CrossRef CAS.
- S. Cornelius, M. Vinnichenko, N. Shevchenko, A. Rogozin, A. Kolitsch and W. Möller, Appl. Phys. Lett., 2009, 94, 042103 CrossRef.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2007, 91, 1529 CrossRef CAS.
- K. Ellmer, J. Phys. D: Appl. Phys., 2001, 34, 3097 CrossRef CAS.
- H. C. Park, D. Byun, B. Angadi, D. Hee Park, W. K. Choi, J. W. Choi and Y. S. Jung, J. Appl. Phys., 2007, 102, 073114 CrossRef.
- M. Gabas, P. Diaz-Carrasco, F. Agullo-Rueda, P. Herrero, A. R. Landa-Canovas and J. R. Ramos-Barrado, Sol. Energy Mater. Sol. Cells, 2011, 95, 2327 CrossRef CAS.
- D. H. Chi, L. T. Thanh Binh, N. Thanh Binh, L. D. Khanh and N. N. Long, Appl. Surf. Sci., 2006, 252, 2770 CrossRef CAS.
- Z. L. Tseng, P. C. Kao, C. S. Yang, Y. D. Juang, Y. M. Kuo and S. Y. Chu, J. Electrochem. Soc., 2011, 158, J133 CrossRef CAS.
- Y. M. Lu, C. M. Chang, S. I. Tsai and T. S. Wey, Thin Solid Films, 2004, 447–448, 56 CrossRef CAS.
- H. P. Chang, F. H. Wang, J. C. Chao, C. C. Huang and H. W. Liu, Curr. Appl. Phys., 2011, 11, S185 CrossRef.
- F. H. Wang, H. P. Chang and J. C. Chao, Thin Solid Films, 2011, 519, 5178 CrossRef CAS.
- J. L. Chung, J. C. Chen and C. J. Tseng, J. Phys. Chem. Solids, 2008, 69, 535 CrossRef CAS.
- J. J. Lu, Y. M. Lu, S. I. Tasi, T. L. Hsiung, H. P. Wang and L. Y. Jang, Opt. Mater., 2007, 29, 1548 CrossRef CAS.
- Y. R. Park and K. J. Kim, Solid State Commun., 2002, 123, 147 CrossRef CAS.
- D. Qian, L. Gerward and J. Z. Jiang, J. Mater. Sci., 2004, 39, 5389 CrossRef CAS.
- Y. Guim, Sh. Li and Ch. Li, Microelectron. J., 2008, 39, 1120 CrossRef.
- M. R. Vaezi, J. Mater. Process. Technol., 2008, 205, 332 CrossRef CAS.
- F. Grasset, G. Starukh, L. Spanhel, S. Ababou-Girard, D. S. Su and A. Klein, Adv. Mater., 2005, 17, 294 CrossRef CAS.
- A. Dodd, A. McKinley, T. Tsuzuki and M. Saunders, J. Alloys Compd., 2010, 489, L17 CrossRef CAS.
- J. S. Jung, Y. H. Kim, S. K. Gi and D. H. Kangl, J. Electroceram., 2009, 23, 272 CrossRef CAS.
- Y. C. Lee, Y. L. Huang, W. H. Lee and F. S. Shieu, Thin Solid Films, 2010, 518, 7366 CrossRef CAS.
- L. Shi, H. Shen, L. Jiang and X. Li, Mater. Lett., 2007, 61, 4735 CrossRef CAS.
- Z. Liul, D. Zhou, S. Gong and H. Li, J. Alloys Compd., 2009, 475, 840 CrossRef.
- N. T. Nolan, Chem. Mater., 2011, 23, 1496 CAS.
- T. Ivanova, A. Harizanova, T. Koutzarova and B. Vertruyen, J. Non-Cryst. Solids, 2011, 357, 2840 CrossRef CAS.
- N. I. Kovtyukhova, E. V. Buzaneva, C. C. Waraksa, B. R. Martin and T. E. Mallouk, Chem. Mater., 2000, 12, 383 CrossRef CAS.
- Z. Yong, T. Liu, T. Uruga, H. Tanida, D. Qi, A. Rusydi and A. T. S. Wee, Materials, 2010, 3, 3642 CrossRef CAS.
- E. Forniés, R. Escobar Galindo, O. Sánchez and J. M. Albella, Surf. Coat. Technol., 2006, 200, 6047 CrossRef.
- A. Climent-Font, F. Pászti, G. García, M. T. Fernández-Jiménez and F. Agulló, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 219–220, 400 CrossRef CAS.
- E. Kotai, Nucl. Instrum. Methods Phys. Res., Sect. B, 1994, 85, 588 CrossRef CAS.
- R. Brydson, L. A. J. Garvie, A. J. Craven, H. Sauer, F. Hofer and G. Cressey, J. Phys.: Condens. Matter, 1993, 5, 9379 CrossRef CAS.
- G. Nehme, R. Mourhatch and P. B. Aswath, Wear, 2010, 268, 1129 CrossRef CAS.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter, 1990, 41, 928 CrossRef CAS.
- R. Revel, D. Bazin and A. M. Flank, J. Synchrotron Radiat., 1999, 6, 717 CrossRef CAS.
- W. Sigle, Annu. Rev. Mater. Res., 2005, 35, 239 CrossRef CAS.
- R. Brydson, H. Sauer, W. Engel and F. Hofer, J. Phys.: Condens. Matter, 1992, 4, 3429 CrossRef CAS.
- A. I. Sheinkman, F. P. Sheinkman, I. P. Dobrovol'skii and G. R. Zvyagina, Izv. Akad. Nauk SSSR Neorg. Mater., 1977, 13, 1447 CAS.
- J. M. Charnock, C. M. B. Henderson, J. F. W. Mosselmans and R. A. D. Pattrick, Phys. Chem. Miner., 2002, 29, 32 CrossRef.
- J.-H. Guo, L. Vayssieres, C. Persson, R. Ahuja, B. Johansson and J. Nordgren, J. Phys.: Condens. Matter, 2002, 14, 6969 CrossRef CAS.
- M. Jullien, D. Horwat, F. Manzeh, R. Escobar Galindo, Ph. Bauer, J. F. Pierson and J. L. Endrino, Sol. Energy Mater. Sol. Cells, 2011, 95, 2341 CrossRef CAS.
- L. W. Chang, Y. C. Sung, J. W. Yeh and H. C. Shih, J. Appl. Phys., 2011, 109, 074318 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |