Nano LiMn2O4 with spherical morphology synthesized by a molten salt method as cathodes for lithium ion batteries
Xuan
Zhao
abcd,
M. V.
Reddy
b,
Hanxing
Liu
*a,
S.
Ramakrishna
c,
G. V. Subba
Rao
b and
B. V. R.
Chowdari
*b
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, 122 Luo Shi Road, Wuhan, 430070, China. E-mail: lhxhp@whut.edu.cn; Fax: +86-27-87651779; Tel: +86-27-87653330
bDepartment of Physics, National University of Singapore, 117542, Singapore. E-mail: phychowd@nus.edu.sg; Fax: +65-67776126; Tel: +65-65162956
cNational University of Singapore Nanoscience and Nanotechnology Initiative (NUSNNI), National University of Singapore, 117576, Singapore
dSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore
First published on 16th July 2012
Abstract
The compound, LiMn2O4 is synthesized by a one-pot molten salt method using NaCl–KCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eutectic melt at various temperatures (T) from 700 to 850 °C and characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Rietveld refinement, surface area and density methods. SEM showed that all the spinel phases LiMn2O4 consist of 1–5 μm sized spherical particles, each of which is composed of ∼50 nm nano-sized aggregates. TEM images show spherical particles with hollow type morphology when the synthesis T is above 800 °C. The cubic lattice parameter, 8.235 (±0.002) Å did not vary much with the T in the range 700–850 °C, which is proven by the similar Mn3+ and Mn4+ amount through XPS results, whereas the surface area varied from 15.6 to 10.3 m2 g−1. The cyclic voltammograms showed the characteristic two-step redox peaks at 3.9/4.1 and 4.1/4.2 V vs. Li for all the compounds in agreement with literature reports. Galvanostatic cycling studies were carried out in the range, 3.5 to 4.3 V vs. Li showed that the LiMn2O4 prepared at 800 °C has the highest discharge capacity of 124 mAh g−1 at second cycle at 0.25 C-rate, and it showed a capacity retention of 96% at 1 C, 2 C and 5 C-rates at the end of 50 cycles. Long-term cycling at 2 C-rate, up to 700 cycles showed a capacity retention of 81%. Thus, LiMn2O4 obtained at 800 °C with uniform hollow spherical particles shows the best electrochemical properties. Complementary electrochemical impedance spectroscopy (EIS) and galvanostatic intermittent titration technique (GITT) studies were carried out and the apparent Li-ion diffusion coefficients (DLi+) were calculated as a function of the applied voltage. The DLi+ values from GITT range from ∼0.1 to 5 × 10−10 cm2 s−1. The values decrease with an increase in the applied voltage and show two minima in good agreement with the available literature data.
:1) as the eutectic melt at various temperatures (T) from 700 to 850 °C and characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Rietveld refinement, surface area and density methods. SEM showed that all the spinel phases LiMn2O4 consist of 1–5 μm sized spherical particles, each of which is composed of ∼50 nm nano-sized aggregates. TEM images show spherical particles with hollow type morphology when the synthesis T is above 800 °C. The cubic lattice parameter, 8.235 (±0.002) Å did not vary much with the T in the range 700–850 °C, which is proven by the similar Mn3+ and Mn4+ amount through XPS results, whereas the surface area varied from 15.6 to 10.3 m2 g−1. The cyclic voltammograms showed the characteristic two-step redox peaks at 3.9/4.1 and 4.1/4.2 V vs. Li for all the compounds in agreement with literature reports. Galvanostatic cycling studies were carried out in the range, 3.5 to 4.3 V vs. Li showed that the LiMn2O4 prepared at 800 °C has the highest discharge capacity of 124 mAh g−1 at second cycle at 0.25 C-rate, and it showed a capacity retention of 96% at 1 C, 2 C and 5 C-rates at the end of 50 cycles. Long-term cycling at 2 C-rate, up to 700 cycles showed a capacity retention of 81%. Thus, LiMn2O4 obtained at 800 °C with uniform hollow spherical particles shows the best electrochemical properties. Complementary electrochemical impedance spectroscopy (EIS) and galvanostatic intermittent titration technique (GITT) studies were carried out and the apparent Li-ion diffusion coefficients (DLi+) were calculated as a function of the applied voltage. The DLi+ values from GITT range from ∼0.1 to 5 × 10−10 cm2 s−1. The values decrease with an increase in the applied voltage and show two minima in good agreement with the available literature data.
Introduction
Lithium mangenese oxide, LiMn2O4 with the spinel structure, is a cheap and eco-friendly alternative cathode material for use in high-power lithium ion batteries and has been researched for more than 20 years.1–7 It is a 4 V cathode and shows a reversible capacity of 100–125 mAh g−1, when cycled between 3.0 and 4.3 V vs. Li, and these capacity values compare well with the practical capacity of 130–135 mAh g−1 shown by the other well-known 4 V cathode, LiCoO2. However, LiMn2O4 suffers from the disadvantage of capacity fading on cycling, especially at temperatures, T ≥ 50 °C. This is ascribed to Mn-dissolution in to the electrolyte, induced by the disproportionation of the Mn3+ ion in to Mn2+ and Mn4+ ions at the particle surface, thereby leading to the loss of active material in the electrode. Several strategies have been adopted to reduce the capacity fading, which include substitution at the Mn-site by other metal ions, like, Li, Al, Co, and surface-coating of the particles of LiMn2O4 by Al, Zr oxides and by metal fluorides.Recently, the methodology of nanotechnology has been adopted to improve the Li-cyclability of LiMn2O4. Thus, Jiao et al.8 have shown that the nano-size, ordered mesoporous Li-excess composition, Li1.12Mn1.88O4 prepared by a hard templating technique gives a capacity of ∼70 mAh g−1 stable up to 100 cycles when cycled between 3.0 and 4.3 V vs. Li, at the high current of 3000 mA g−1 (current rate, 30 C) at ambient temperature. Further, at 50 °C and at a current rate of 0.3 C, ∼98% of the initial capacity was retained up to 100 cycles. Similarly, Lee et al.9 found that ultrathin nano-wires of LiMn2O4 (10 nm diameter and several μm in length) prepared by solvothermal reaction, followed by solid-state lithiation, can deliver capacities of 100 and 78 mAh g−1 at very high current rates of 60 C and 150 C, respectively in a larger potential window with good capacity retention and outstanding structural stability. Thus, the morphology and nano-size LiMn2O4 with high crystallinity do have a significant impact on its Li-cyclability and suppression of capacity fading.10
A large number and wide variety of methods have been adopted for the synthesis of LiMn2O4 and these include direct solid state reaction of the constituent oxides or carbonates/nitrates, metal–organic and/or polymer precursors etc..4–6 Of late, however, molten salt method (MSM) has been receiving attention due to several advantages: 1. it is a one-pot method where the constituent reactants directly yield the desired phase. By a judicious choice of the molten salt, and control of temperature and time of reaction, it is possible to obtain the LiMn2O4 as a nano-phase material. 2. By using an eutectic melt of the salt-mixture, it is possible to synthesize LiMn2O4 at a much lower temperature than that is required in other methods. 3. The MSM is amenable to large-scale synthesis and the salt(s) can be recovered for re-use, thus making it a cost-effective method.
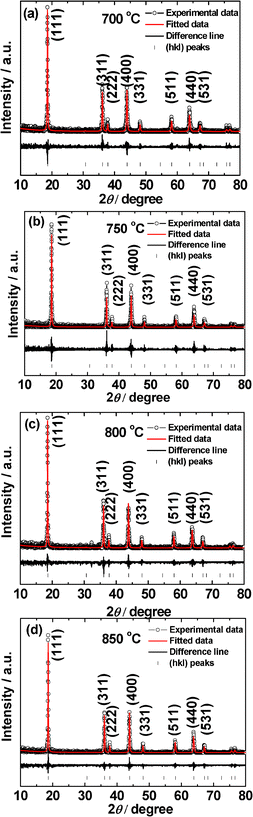 | ||
| Fig. 1 X-ray diffraction patterns of LiMn2O4 prepared at different temperatures (a) 700 °C, (b) 750 °C, (c) 800 °C and (d) 850 °C. Fitted patterns, difference lines and Miller indices (hkl) are shown. Cu–Kα radiation. | ||
In the literature, there are a few reports on the preparation of LiMn2O4 using LiCl, NaCl, KCl and CaCl2 as molten salt or eutectic melts.11–14 Some of the methods used a two step processes, like post-annealing in air at 750 °C for 4 h to get fine crystallinity.11 However, from the literature, it is easy to conclude that, LiCl molten salt favors the particle growth, while using NaCl and KCl molten salts one can obtain LiMn2O4 particles at nano-sized level. The LiMn2O4 prepared using KCl as the molten salt, showed a few hundred nanometer-sized particles with irregular aggregation. Besides, the electrochemical performance of LiMn2O4 synthesized using NaCl is better than that synthesized using LiCl as the molten salt, but inferior to the compound prepared using KCl as the molten salt.13
Based on our previous experience on molten salt synthesis of various mixed oxides, we concluded that the chloride salts have a beneficial role in the synthesis of LiMn2O4, when compared with nitrate based molten salts. In this work, a NaCl–KCl (1:1) eutectic mixture is used to prepare nano-phase LiMn2O4. The eutectic mixture has a melting point of 650 °C, which is lower than those of KCl and NaCl at 774 °C and 801 °C, respectively. The structure, morphology and electrochemical properties are described and discussed.
Experimental
The LiMn2O4 compounds were synthesized using MnCO3·xH2O (Merck, Mn% = 43%–46%), Li2CO3 (Merck, 99%), NaCl (Merck, 99.5%) and KCl (Merck, 99.5%) as starting materials. Stoichiometric amounts of Mn- and Li- carbonate (molar ratio 2:1) along with 0.5 M NaCl–0.5 M KCl were thoroughly mixed in an agate mortar. The molar ratio of total metal ions to the eutectic salt was fixed at 1:6. The mixture was transferred into a recrystallized alumina crucible (50 mL) and heated in a box furnace (Carbolite, UK) at temperatures 700, 750, 800, 850 °C in air for 6 h each at a heating rate of 3 °C min−1, and finally cooled to room temperature by furnace shut-off. The obtained four batches of LiMn2O4 samples were washed several times with de-ionized water to remove the Na and K-salts, filtered, washed with ethanol and finally dried at 80 °C for 12 h in air. The compounds were stored in a desiccator.
The crystal structure was identified by X-ray diffraction (XRD) (Philips X'PERT MPD unit with Cu–Kα radiation). The unit-cell lattice parameters were obtained by Rietveld refinement of the powder XRD data using the software TOPAS Version 2.1. The morphology and structure of the samples was examined by scanning electron microscope (SEM) (JEOL JSM-6700F) and transmission electron microscopy (TEM, JEOL 3010, operated at 300 kV for the compounds prepared at 700, 750, and 800 °C and TEM, JEOL 2010, operated at 200 kV used for the compound prepared at 850 °C). The oxidation states of the transition metal species were studied by X-ray photoelectron spectrometry, Kratos AXIS UltraDLD (Kratos Analytical Ltd., working pressure, 5 × 10−9 Torr, X-ray source, Mono Al Kα hν = 1486.71 eV, 5 mA, 15 kV). The energy scale was adjusted based on the graphite peak in the C 1 s spectrum at 284.5 eV. The Tristar 3000 (Micromeritics, USA) and AccuPyc 1330 pycnometer (Micromeritics, USA) were used to obtain the BET surface area and density of the compounds, respectively.
The electrochemical measurements were carried out using a CR-2016-type coin cell with a Celgard 2502 membrane separator, and 1 M LiPF6-ethylene carbonate (EC)–diethyl carbonate (DEC) (1:1 in volume; Merck) as electrolyte. The composite cathode electrode was composed of 70 wt.% active material, 15 wt.% conductive carbon black (Super P) and 15 wt.% Kynar binder, which were mixed in N-methyl pyrrolidinone (NMP) solvent to dissolve the binder and to obtain viscous slurry. The slurry was then coated on to an etched aluminum foil (20 μm thickness) using the doctor blade method.
 | ||
| Fig. 2 SEM images of LiMn2O4 compounds prepared at different temperatures (a) 700 °C, (b) 750 °C, (c) 800 °C, and (d) 850 °C. The scale bars are 1 μm for all the compounds. | ||
The electrode foil was dried in an oven at 100 °C for 10 h to evaporate NMP and it was then pressed between stainless steel rollers to ensure good electrical contact between the Al-foil current collector and the active material of the composite. The foil was then cut into 16 mm circular disks. Geometrical area of the electrode was 2.0 cm2 and weight of the active material was 4–5 mg. Cells were assembled in Argon-filled glove box (MBraun, Germany) using Li-metal foil (Kyokuto Metal Co., Ltd) as counter (anode) and reference electrode. Cyclic voltammetry studies were carried using computer controlled Mac-pile II system (Bio-logic, France). Charge–discharge cycling between 3.5–4.3 V vs. Li at room temperature was carried out using bitrode (Model SCN, Bitrode, USA), and galvanostatic intermittent titration technique (GITT) was carried out using Arbin Battery Tester (BT2043, USA). Electrochemical impedance spectroscopy (EIS) was carried on cells using Solartron Impedance/gain-phase Analyzer (model SI 1255) coupled with a potentiostat (SI 1268) at room temperature. The frequency was varied from 0.18 MHz to 3 mHz with an alternating voltage signal amplitude of 10 mV.
Results and discussion
Physical characterization
The as-prepared LiMn2O4 compounds are free-flowing black crystalline powders. The Rietveld refined XRD patterns of the compounds prepared at various temperatures (700–850 °C) are shown in Fig. 1. All the XRD patterns can be indexed to the well-known LiMn2O4 spinel structure1–4 with the space group, Fd-3m (Space Group: 227). No impurity phases were detected. All the diffraction peaks are sharp and intense regardless of synthesis temperature (T), which is an indication of good crystallinity of samples. This can be attributed to the NaCl–KCl eutectic mixture used, which provides liquid environment at T > 650 °C and enables the Mn and Li-salt dissolution, oxidation of the Mn-ion to Mn3+ and Mn4+ ions and nucleation followed by the formation of nano-phase LiMn2O4 at T ≥ 700 °C.The cubic lattice parameter (a) of LiMn2O4 synthesized at different temperatures (T) are given in Table 1. The a of LiMn2O4 is 8.235 (±0.002) Å when the T ≥ 750 °C. The slightly increasing of the a value when compared to T = 700 °C is because of the slightly increasing of Mn3+ in the compounds.15 The a value of 8.235 (±0.002) Å matches well with the value of 8.236 Å of the LiMn2O4 given in the JCPDS-no. 88-1026.
| Synthesis temperature T (°C) | Lattice parameter (±0.001) (Å) | Experimental/thoretical density (±0.002) (g cm−3) | BET surface area (±0.1) (m2 g−1) | Crystallite size (±0.1) (nm) |
|---|---|---|---|---|
| 700 | 8.230 | 4.306/(4.309) | 15.6 | 58.8 |
| 750 | 8.236 | 4.252/(4.300) | 15.9 | 71.5 |
| 800 | 8.234 | 4.449/(4.303) | 13.6 | 87.0 |
| 850 | 8.237 | 4.231/(4.299) | 10.3 | 79.5 |
Fig. 2a–d show SEM images of LiMn2O4 synthesized at different temperatures. As can be seen, the particle size and the morphology of LiMn2O4 prepared at different temperatures do not vary much: all compounds are composed of 1–5 μm sized spherical particles with ∼50 nm nano-sized aggregates. This type of interesting morphology is not obtained in LiMn2O4 prepared by MSM using other kinds of molten salts.11–14 The reason may be due to the differences in the eutectic temperature of melt (650 °C for NaCl–KCl, 1:1), the reactivity of molten salt in effectively dissolving the Li- and Mn-ions and the combined effect of cations, Na and K in the molten salt. Moreover, the spherical morphology has resulted in a slightly higher BET surface area (10–15 m2 g−1) in spite of the synthesis temperature (T) being ≥ 700 °C (Table 1). As expected, the BET surface area decreases as the T increases, from 15.6 (700 °C) to 10.3 (850 °C) m2 g−1 as the nano-sized aggregates size increasing with T (Fig. 2 & 3).
Fig. 3a–d show the TEM images of LiMn2O4 synthesized at different T. At 700 °C, the nano-sized particles gather together, but no obvious spherical second-particles formed. At 750 °C, the nano-sized particles gathered densely. When the temperature is as high as 800 °C and 850 °C, there are more spherical, second-particles are formed with hollow type spherical morphology. The size of the spherical second-particles are more uniform when the LiMn2O4 was synthesized at 800 °C. High resolution (HR)TEM image of the LiMn2O4 synthesized at 800 °C is shown in Fig. 3e, and it showed a d value ≈ 0.21 nm, which corresponds to the (400) plane (hkl) of LiMn2O4. Besides, the SAED pattern of the same compound (Fig. 3f) is indexed well with the spinel structure of the LiMn2O4. TEM images prove the good crystallinity of the sample.
 | ||
| Fig. 3 TEM images of LiMn2O4 compounds prepared at different temperatures, (a) 700 °C, (b) 750 °C, (c) 800 °C, and (d) 850 °C, (e) 800 °C, high resolution lattice image, (f) SAED pattern of LiMn2O4 synthesized at 800 °C. | ||
X-ray photoelectron spectroscopy was carried out to find the manganese valency. The core level experimental spectra for Mn 2p of LiMn2O4 synthesized at different temperatures are shown in Fig. 4a. There are no obvious peak shifts in binding energy values with preparation temperature, which indicates the manganese oxidation state in all these four samples are same and further proves the similar lattice parameter a values in the XRD results. For clarity, core level experimental and fitted spectra of the LiMn2O4 synthesized at 800 °C are shown in Fig. 4b. The two peaks located at 641.5 (±0.2) eV and 642.8 (±0.2) eV corresponds to Mn3+ and Mn4+, respectively.16 The total peak area of Mn3+ peak is slightly higher than that of the Mn4+, which indicates the slightly higher content of the Mn3+ amount in the compound.
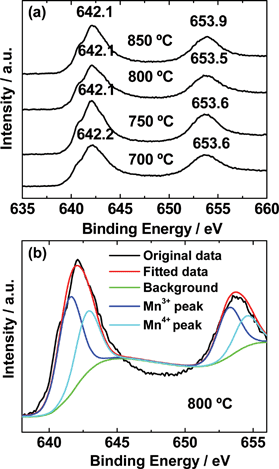 | ||
| Fig. 4 (a) Mn 2p3/2 and Mn 2p1/2 XPS spectra of LiMn2O4 synthesized at different temperatures, (b) peak fitted spectra of the LiMn2O4 synthesized at 800 °C. | ||
Cyclic voltammetry
The second-cycle cyclic voltammograms (CV) of Li–LiMn2O4 cells in the voltage range of 3.5 to 4.3 V at the slow scan rate of 58 μV s−1 are shown in Fig. 5. The Li-metal was the counter and reference electrode. Two typical redox peaks around 3.9/4.1 V and 4.0/4.2 V vs. Li are shown for all the LiMn2O4 synthesized at various temperatures. The two anodic peaks at ∼4.0 V and ∼4.2 V are corresponding to Li-extraction from the 8 a sites in the crystal lattice, and the formation of Li0.5Mn2O4 and Li∼0.25Mn2O4, respectively.1,15 LiMn2O4 prepared at 800 °C shows relatively sharp and high-intensity peak currents and low voltage hysteresis between the cathodic and anodic peak potentials, which reflects good reversibility between charge and discharge processes. The low intensity cathodic peak, appearing as a shoulder peak below 3.9 V can be attributed to the ‘formation cycle’ of the electrode material during initial cycles whereby minor structural rearrangement of the lattice takes place.1,15,17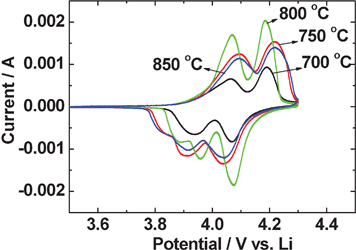 | ||
| Fig. 5 Cyclic voltammograms (CV) of LiMn2O4 prepared at different temperatures. The 2nd cycle CV are shown. Scan rate: 58 μV s−1; Li-metal is the counter and reference electrode. | ||
Galvanostatic cycling
The second cycle galvanostatic charge–discharge profiles for LiMn2O4 prepared at various temperatures are displayed in Fig. 6a. For the LiMn2O4 prepared at 800 °C, the profiles of the 1,2,10 and 50 cycles are shown in Fig. 6b. The cells were cycled at a constant current of 30 mA g−1 (current rate, 0.25 C, assuming 1 C = 120 mA g−1), in the voltage range of 3.5 to 4.3 V vs. Li. As can be seen, all the compounds show characteristic voltage plateaus at 4.0/4.15 V and 3.95/4.1V during charge and discharge, respectively (Fig. 6a), which correspond well to the redox peaks in the CV and in very good agreement with the reports in the literature.1,11–15,17 The charge capacities at the 2nd cycle of LiMn2O4 prepared at 700, 750, 800 and 850 °C are 120, 118, 126 and 114 (±3) mAh g−1, respectively. The corresponding discharge capacities are 110, 115, 124, 112 (±3) mAh g−1, respectively. The LiMn2O4 compound synthesized at 800 °C shows the best electrochemical reversibility. The capacity vs. cycle number plots of LiMn2O4 up to 50 cycles are shown in Fig. 7a. The capacity retention of all the compounds prepared at various temperatures are 91%–92% (between the 2nd and the 50th cycle), which are better than the LiMn2O4 synthesized at similar temperatures by other methods, for example using LiCl as the molten salt.11 The LiMn2O4 prepared at 800 °C shows slightly higher discharge capacity of 113 mAh g−1 after 50 cycles (91% capacity retention).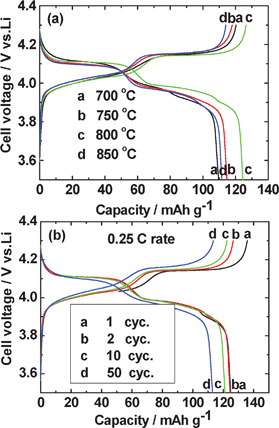 | ||
| Fig. 6 (a) Galvanostatic charge and discharge profiles (second cycle) of LiMn2O4 prepared at different temperatures. (b) Galvanostatic charge and discharge capacity profiles (1st, 2nd, 10th and 50th cycle) of LiMn2O4 prepared at 800 °C. Voltage range: 3.5–4.3 V vs. Li; current: 30 mA g−1 (0.25 C). | ||
The C-rate capabilities were studied on the LiMn2O4 synthesized at 800 °C at a current rate of 1 C, 2 C, 5 C and 10 C-rate (assuming 1 C = 120 mA g−1). The charge–discharge cycling curves (not shown) are similar to those recorded at 30 mA g−1 (0.25 C). A slight polarization in the charge–discharge voltage are seen. The capacity vs. cycle number plots (Fig. 7b) indicate a 2nd cycle discharge capacity of 119 mAh g−1, 121 mAh g−1, 98 mAh g−1 and 75 mAh g−1 at 1 C, 2 C, 5 C and 10 C-rate, respectively. The capacity retention is 96% after 50 cycles at 1 C, 2 C and 5 C-rates, which are better than the capacity retention of LiMn2O4 cycled at the lower current rate of 0.25 C. When the current is increased to 10 C-rate, the capacity retention is 96% between 16 to 50 cycles. The fast capacity fading during the first 15 cycles maybe due to the large current rate (10 C) and incomplete formation cycle. The stable rate capacity retention is because of the spherical particles with nano-aggregates morphology of the compound.
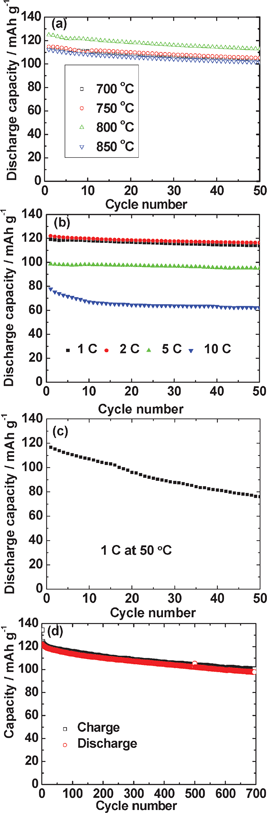 | ||
| Fig. 7 (a) Discharge capacity vs. cycle number plots of LiMn2O4 synthesized at different temperatures (700 to 850 °C). Voltage range, 3.5–4.3 V at a current of 30 mA g−1(0.25 C). (b) Discharge capacity vs. cycle number plots of LiMn2O4 synthesized at 800 °C at 1 C, 2 C, 5C and 10 C-rate up to 50 cycles. Voltage range: 3.5–4.3V vs. Li (1 C = 120 mA g−1). (c) LiMn2O4 synthesized at 800 °C cycled at 1 C-rate at elevated temperature. (d) Upto 700 cycles; 2 C-rate. | ||
The capacity retention of the LiMn2O4 synthesized at 800 °C at a current rate of 1 C was also tested at 50 °C (Fig. 7c). The initial discharge capacity is 117 mAh g−1. After 50 cycles, it showed a capacity of 76 mAh g−1 (capacity retention is 65% between 2–50 cycles), which is higher than that reported by others on undoped or uncoated LiMn2O4 at similar testing temperature. Wang et al.18 reported that the undoped LiMn2O4 has a discharge capacity retention less than 45% after 40 cycles at 55 °C. In the research of Qing et al.,19 the uncoated LiMn2O4 has a capacity retention around 42% after 50 cycles at 55 °C. The capacity retention of pristine LiMn2O4 cycled after 27 cycles at 1 C-rate (1 C = 100 mAh g−1) at 55 °C is only 30% as reported by Yu et al.20 The superior capacity retention at elevated temperature in the present study can be explained by the high crystallinity of the compound as well as the existing of the large secondary particles (spherical particles).
Long term cycling was carried out at 2 C-rate for the LiMn2O4 synthesized at 800 °C: the charge and discharge capacities are 100 and 98 mAh g−1 at the end of 700 cycles. The discharge capacity retention is 81% after 700 cycles proving very good cathodic performance at 2 C-rate (Fig.7d). There are only a few reports of long-term cycling of LiMn2O4 in the literature: Shaju and Bruce21 reported cycling data of stoichiometric nano LiMn2O4 prepared by the sol–gel technique (I) and by resorcinol–formaldehyde gel decomposition (II). Both methods gave nano-size particles which are fused together, and in addition, the second method gave rise to the phase with a porous morphology, with pores between 2–5 μm. When cycled at 0.5 C (1 C = 148 mA g−1) in the range, 3.5–4.3 V vs. Li, at 30 °C, LiMn2O4-II gave an initial discharge capacity of 131 mAh g−1 with a capacity retention of 90% at the end of 200 cycles. At 10 C-rate, LiMn2O4-II showed an initial discharge capacity of 120 mAh g−1 with a capacity retention of 91% at the end of 1000 cycles. At 0.5 C, under the same cycling conditions, LiMn2O4-I gave an initial discharge capacity of 133 mAh g−1 with a capacity retention of only 60% at the end of 200 cycles. Thus, it is concluded that the long-term cycling performance of MSM LiMn2O4 prepared at T = 800 °C compares very well with the literature data.
Impedance spectral studies
The electrochemical impedance spectra (EIS) were recorded on the LiMn2O4 synthesized at different temperatures. The counter electrode was Li-metal. The Nyquist plots (Z′ vs.−Z′′) are shown in Fig. 8. The measurements were done at open circuit voltage (OCV ∼3 V) conditions. The impedance spectra of all compounds consist of one clear semicircle in the high-frequency range and a straight line with an inclined slope in the medium frequency range. The semicircle does not touch the Z′-axis indicating a slightly non-ideal behavior due to the composite nature of the electrode. The semicircle diameter indicates the surface film resistance (Rsf) since the cells are in the OCV condition.22–25 The Rsf values are 204, 168, 162 and 291 (±5) Ω, respectively for LiMn2O4 prepared at 700, 750, 800 and 850 °C. It is clear that LiMn2O4 prepared at 800 °C has the smallest value of Rsf and this is consistent with the observation that the LiMn2O4 synthesized at 800 °C has largest discharge capacity and very good cycling performance.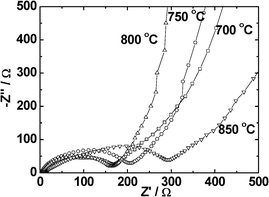 | ||
| Fig. 8 Impedance spectral Nyquist plots (Z′ vs. −Z′′) measured at open circle voltage (OCV ∼3.0 V vs. Li) of LiMn2O4 synthesized at different temperatures. | ||
GITT study and DLi+ calculation
Galvanostatic intermittent titration technique (GITT) is a technique to obtain the apparent Li-diffusion coefficient (DLi+) of LiMn2O4 and is presumed to be an accurate method.23,26–28 GITT studies were carried out on LiMn2O4 compound synthesized at 800 °C from 3.5 to 4.3 V in both charge and discharge processes. The cell was charged and discharged at a constant current rate of 0.25 C (30 mA g−1) for 10 min, then a relaxation time of 40 min was applied to the cell. The full GITT processes are shown in Fig. 9a. The applied current flux for a time, τ (10 min) and the resulting voltage profile is shown in Fig. 9b. The DLi+ at different voltages are calculated based on the eqn (1):23,26,27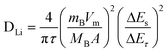 | (1) |
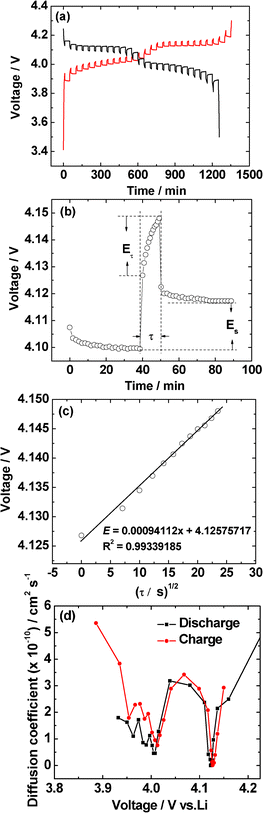 | ||
| Fig. 9 (a) GITT curves of LiMn2O4 synthesized at 800 °C during the second cycle in the voltage of 3.5–4.3 V. (b) GITT curve at 4.1 V with schematic labeling of different parameters. (c) Plot of voltage against τ1/2 to show the linear fit. (d) The Li-ion diffusion coefficients (DLi+) calculated from the GITT data during both charge and discharge processes. | ||
In the above equation, ΔEs is the change in the steady state voltage and ΔEτ is the total change in cell voltage as shown in Fig. 9b. The term mB is the active mass (LiMn2O4) in the electrode, MB the formula mass of LiMn2O4, Vm is the molar volume of the LiMn2O4, and A is the geometrical area of the electrode (2 cm2). This equation only can be applied when the variation of cell voltage during titration is found to show a straight line behavior on plotting against τ1/2. Fig. 9c clearly shows the straight line trend of the voltage as a function of τ1/2, the R2 value is 0.99, which is very close to 1. The calculated DLi+ from GITT data are shown in Fig. 9d and the values range from ∼0.1 to 5.5 × 10−10 cm2 s−1. The DLi+vs. voltage of both charge and discharge processes have two minima near 4.0 V and 4.12 V which corresponding to the two main redox processes noted in the CV studies (Fig. 5).
It is pertinent to compare the presently observed DLi+ values with those reported in the literature: Shaju et al.23 determined the DLi+ values on micro-crystalline LiMn2O4 by GITT and they were found to be in the range, 1 to 9 × 10−10 cm2 s−1 depending on the voltage in good agreement with the present study. Xie et al.27 determined the DLi+ values on pulsed laser deposited (PLD) thin films of LiMn2O4 by EIS and GITT and found the values to be in the range, ∼1 × 10−13 to 10−11 cm2 s−1 and ∼1 × 10−14 to 10−11 cm2 s−1, respectively, depending on the voltage. Several earlier reports observed DLi+ values (ranging from ∼1 × 10−12 to 10−10 cm2 s−1) on thin films of LiMn2O4, but in all cases, and by both the EIS and GITT methods, the DLi+ values were found to decrease with an increase in the voltage. Thus, it is concluded that the present DLi+ values on nano-size hollow spherical LiMn2O4 are slightly larger, as can be expected, than those reported on the thin films of LiMn2O4.
Conclusions
LiMn2O4 phases were synthesized by a one-pot molten salt method (MSM) using NaCl–KCl (1:1) as eutectic melt. Spinel phases are obtained when the synthesis temperature (T) was varied from 700 to 850 °C. SEM showed that all the LiMn2O4 phases consist of 1–5 μm sized spherical particles composed of ∼50 nm nano-sized aggregates. TEM images reveal the spherical particles are hollow when the synthesis temperature is above 800 °C. This kind of morphology is expected as the best way to improve both rate capability and electrode density.28
In the experiment, the cubic lattice parameter, 8.235 (±0.002) Å did not vary much with the T in the range 750–850 °C, which is also proved by the similar Mn3+ and Mn4+ amount through XPS results. As a result, the main difference among the samples synthesized at different temperatures is the morphology. LiMn2O4 obtained at 800 °C with uniform hollow spherical particles has the best electrochemical properties. The cyclic voltammograms showed the characteristic two-step redox peaks at 3.9/4.1 and 4.0/4.2 V vs. Li in agreement with literature reports. The galvanostatic cycling studies between 3.5 and 4.3 V vs. Li showed that the LiMn2O4 prepared at 800 °C has the highest discharge capacity of 124 mAh g−1 at second cycle at 0.25 C-rate, and it showed a capacity retention of 96% at 1 C, 2 C and 5 C-rates (range, 2 to 50 cycles). Long-term cycling at 2 C-rate, up to 700 cycles showed a capacity- retention of 81%. Complementary EIS and GITT study was carried out and the apparent Li-ion diffusion coefficients (DLi+) were estimated as a function of the applied voltage. The DLi+ values, ∼0.1 to 5 × 10−10 cm2 s−1 at various voltages from GITT data agree well with those reported on micro-crystalline LiMn2O4 but are larger when compared to those DLi+ reported by GITT on thin films of LiMn2O4. It is concluded that the NaCl–KCl (1:1) molten salt method is favored to synthesize spinel LiMn2O4 with hollow spherical particles comprised of nano-sized aggregates. This kind of morphology improves the electrochemical performance of the LiMn2O4.
Acknowledgements
One of the authors (XZ) is thankful to the Chinese Research Council, China for financial support during her stay at the National University of Singapore and Prof. Subodh Mhaisalkar at School of Materials Science and Engineering, Nanyang Technological University for his support. The authors (XZ & MVR) thank the Ministry of Education (MOE), Singapore, for support under the research grant No. R-284-000-076-112, National Research foundation (NRF) Singapore, NRF-CRP-4-2008-03, NRF-CRP Grant No R-144-000-295-281 and R-143-000-360-281.References
- G. Amatucci and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, K31 CrossRef CAS.
- W. V. Schalkwijk, B. Scrosati, Advances in lithium-ion batteries, Kluwer Academic/Plenum, New York, 2002 Search PubMed.
- G. A. Nazri, G. Pistoia, Lithium batteries: science and technology, Kluwer Academic, New York, 2003 Search PubMed.
- K. Ozawa Edited, Lithium ion rechargeable batteries: materials, technology, and new applications, Wiley-VCH, Weinheim, 2009 Search PubMed.
- T. Ohzuku and R. J. Brodd, J. Power Sources, 2007, 174, 449 CrossRef CAS.
- Y. G. Xia, Q. Zhang, H. Y. Wang, H. Nakamura, H. Noguchi and M. Yoshio, Electrochim. Acta, 2007, 52, 4708 CrossRef CAS.
- L. F. Nazar, B. L. Ellis and K. T. Lee, Chem. Mater., 2010, 22, 691 CrossRef.
- F. Jiao, J. L. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711 CrossRef CAS.
- H. W. Lee, P. Muralidharan, R. Ruffo, C. M. Mari, Y. Cui and D. K. Kim, Nano Lett., 2010, 10, 3852 CrossRef CAS.
- Q. T. Qu, L. J. Fu, X. Y. Zhan, D. Samuelis, J. Maier, L. Li, S. Tian, Z. H. Li and Y. P. Wu, Energy Environ. Sci., 2011, 4, 3985 CAS.
- W. P. Tang, X. J. Yang, Z. H. Liu, S. Kasaishi and K. Ooi, J. Mater. Chem., 2002, 12, 2991 RSC.
- K. Du, L. Qi, G. R. Hu and Z. D. Peng, Chin. J. Inorg. Chem., 2006, 22, 867 CAS.
- K. Du, Y. N. Yang, G. R. Hu, Z. D. Peng and L. Qi, Chin. J. Inorg. Chem., 2008, 24, 615 CAS.
- M. Helan, L. J. Berchmans and A. Z. Hussain, Ionics, 2010, 16, 227 CrossRef CAS.
- M. V. Reddy, S. S. Manoharan, J. John, B. Singh, G. V. Subba Rao and B. V. R. Chowdari, J. Electrochem. Soc., 2009, 156, A652 CrossRef CAS.
- K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Solid State Ionics, 2002, 152–153, 69 CrossRef CAS.
- K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Solid State Ionics, 2002, 148, 343 CrossRef CAS.
- C. Y. Wang, S. G. Lu, S. R. Kan, J. Pang, W. R. Jin and X. J. Zhang, J. Power Sources, 2009, 189, 607 CrossRef CAS.
- C. B. Qing, Y. Bai, J. M. Yang and W. F. Zhang, Electrochim. Acta, 2011, 56, 6612 CrossRef CAS.
- L. H. Yu, X. P. Qiu, J. Y. Xi, W. T. Zhu and L. Q. Chen, Electrochim. Acta, 2006, 51, 6406 CrossRef CAS.
- K. M. Shaju and P. G. Bruce, Chem. Mater., 2008, 20, 5557 CrossRef CAS.
- M. D. Levi and D. Aurbach, J. Phys. Chem. B, 1997, 101, 4630 CrossRef CAS.
- K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, J. Mater. Chem., 2003, 13, 106 RSC.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Phys. Chem. C, 2007, 111, 11712 CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2010, 195, 5768 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569 CrossRef CAS.
- J. Xie, K. Kohno, T. Matsumura, N. Imanishi, A. Hirano, Y. Takeda and O. Yamamoto, Electrochim. Acta, 2008, 54, 376 CrossRef CAS.
- O. K. Park, Y. H. Cho, S. H. Lee, H.-C. Yoo, H.-K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621 CAS.
| This journal is © The Royal Society of Chemistry 2012 |