Strengthening of degraded cellulosic material using a diamine alkylalkoxysilane
Zied
Souguir
a,
Anne-Laurence
Dupont
*a,
Kateryna
Fatyeyeva
b,
Gérard
Mortha
c,
Hervé
Cheradame
d,
Stéphane
Ipert
e and
Bertrand
Lavédrine
a
aCentre de Recherche sur la Conservation des Collections, Museéum National d'Histoire Naturelle, CNRS USR 3224, 36 rue Geoffroy-Saint-Hilaire, 75005, Paris, France
bLaboratoire Polymères, Biopolymères, Surfaces, CNRS UMR 6270, Université de Rouen, Bd. Maurice de Broglie, 76821 Mont Saint Aignan Cedex, France
cLaboratoire de Génie des Procédés Papetiers (LGP2), UMR CNRS 5518, Grenoble INP-Pagora, 461 rue de la Papeterie, BP65, 38402 Saint Martin d'Hères Cedex, France
dLaboratoire Analyse et Modeélisation pour la Biologie et l'Environnement, CNRS UMR 8587, Universiteé d'Evry, Boulevard François Mitterrand, 91025 Evry cedex, France
eCentre de Conservation du Livre (CCL) L'Enclos Saint-Césaire, Impasse des Mourgues, 13200 Arles, France
First published on 13th June 2012
Abstract
The physicochemical modifications in papers upon introducing AEAPMDMS (3 N-(2-aminoethyl)-3-aminopropylmethyl-dimethoxysilane) as a dry strength and deacidification agent were explored. The double amine functionality was shown to favor penetration in the cellulose fibers, with large uptakes being achieved at low concentration. In situ polymerization in the paper was demonstrated using 29Si CP-MAS and 1H NMR. The distribution of the compound inside and on the fibers' surface was evidenced with SEM-EDS and XPS. The deacidification efficacy was established. The strengthening effect was shown to arise from the interaction of AEAPMDMS with the fibers, and depended on the fiber composition (lignin content) and oxidation state, with a higher efficiency for the less degraded and less lignified fibers. In the most degraded papers, the occurrence of yellowing was interpreted as due to the formation of Schiff bases, where the amine functional group reacts with the carbonyl groups on the oxidized cellulose.
Introduction
The acidity generated during the natural aging of paper is the main cause for its deterioration. Deacidification, a chemical treatment for paper preservation, involves the neutralization of the acids present in the paper and the deposition of an alkaline compound such as calcium carbonate (often referred to as “alkaline reserve”) to prevent, or at least delay, further acidification. Mass scale deacidification processes are commercially available for libraries and archives in several countries1,2 and institutions may resort to them as a prevention action for the collections at risk of acidification on the short or mid term. However for collections which are presently strongly acidic and so brittle that they cannot be handled without risking the loss of material, these mass treatments fail at providing a viable solution as they do not impart any mechanical strengthening. A new chemical process that simultaneously deacidifies, introduces an alkaline reserve, and improves the mechanical properties of the paper by introducing aminoalkylalkoxysilanes has been proposed recently by the authors.3–6 These molecules are used for the formation of hybrid materials, the immobilization of biomolecules or the modification of surface activity.7–10 Alkoxysilanes have been used for the reinforcement of natural fibers. A review of the literature shows that most of these alkoxysilanes are tri- and tetra-functional and that they form interpenetrated networks.11–13 These networks of synthetic polymers with natural fibers can result in composite materials. The experimental conditions (solvent and temperature) to produce these materials however are too drastic to be applied to historic cellulosic documents.The novel solvent phase process solution based on aminoalkylalkoxysilanes (AAASs) targeted for libraries and archive materials provides, besides deacidification and alkaline reserve deposition, a clear improvement in the mechanical properties of the paper, enhances the stability toward aging, and confers fungistatic properties to the paper.3–5,14 The understanding of the chemical and physicochemical events occurring between cellulose macromolecules and AAAS is essential in the interpretation of their effect as strengthening agents for cellulosic materials and in enabling the design of treatment molecules specifically adapted to papers in different alteration (oxidation) states. In our previous work, aminopropylmethyldiethoxysilane (AMDES), which has been thoroughly characterized,6,15 was chosen due to its capability to polycondensate as a linear polymer upon hydrolysis. In that work, besides the capacity of AMDES to improve the mechanical properties of degraded and non degraded papers, the even penetration of the compound in the fiber network was shown. It was determined that the quantity of AMDES required for a significant strengthening efficacy was at least 10% wt/wt. For a mass treatment of paper based items this quantity would benefit from being downsized, were it only for cost-efficiency reasons. In the present work, we studied a different linear AAAS, AEAPMDMS (3 N-(2-aminoethyl)-3-aminopropylmethyl-dimethoxysilane), which has the specificity of bearing two amine functions (primary and secondary). We have demonstrated previously the important role of the amine functional group in favoring the uptake. By allowing for a much larger proton sponge capacity, the double amine functionality was thought to enable a reduction of the quantity of AAAS in the treatment solution while obtaining the same efficacy in terms of uptake and alkaline reserve. The relation between the concentration of AEAPMDMS in the treatment solution and several resulting chemical and physical characteristics of papers, such as the uptake and alkaline reserve but also the mechanical properties (resistance to double folds and resistance to tensile strength in zero-span and in the coaxial configurations) and the color formation were investigated. The penetration of AEAPMDMS into the fibers was studied using scanning electron microscopy and energy dispersive X-ray spectrometry (SEM-EDS) and X-ray photoelectron spectroscopy (XPS).
Experimental
Materials
The paper henceforth called P2 is made of cotton linters (>95%) with traces of softwood pulp, and contains no fillers or sizing (basis weight 76 g m−2, cold extract pH = 6.34). The paper henceforth called P3 is made of groundwood pulp (75%) and softwood cellulose (25%); it contains 20% kaolin filler and is sized with alum-rosin (basis weight 80 g m−2, cold extract pH = 5.4). Both papers were specially made at a paper mill in The Netherlands in 1990 for the STEP project.16 Two naturally aged books, henceforth called B1 (dated 1923, mix of groundwood pulp and softwood chemical pulp, with kaolin fillers, cold extract pH = 5.3) and B2 (dated 1946, 100% thermomechanical or chemi-thermomechanical pulp, no fillers, cold extract pH = 4.7) were also included in the study.The solvent, hexamethyldisiloxane (HMDS), and the AEAPMDMS (Fig. 1) were purchased from ABCR, Gelest.
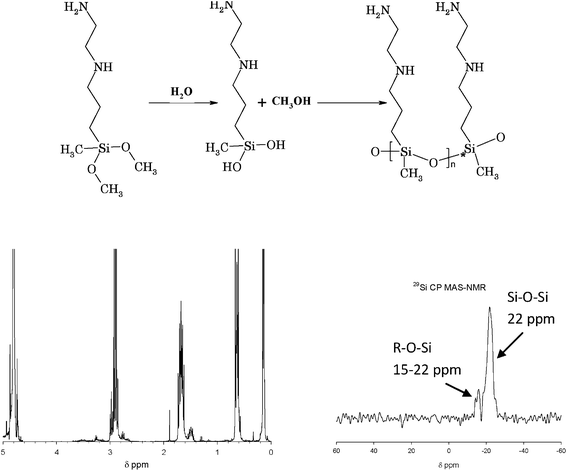 | ||
| Fig. 1 Top: 3 N-(2-aminoethyl)-3-aminopropylmethyl-dimethoxysilane (AEAPMDMS) and its reaction path (hydrolysis and condensation) to form poly-(3 N-(2-aminoethyl)-3-aminopropylmethyl-silanediol). Bottom: A 1H NMR spectrum of a D2O extract (left) and a 29Si CP MAS-NMR spectrum at room temperature (right) of P2 treated with AEAPMDMS (chemical shifts determined using TMS as the internal standard). | ||
A simple impregnation system previously tested ensured the reproducibility of the uptake from sample to sample and from one experiment to the next.6 Four paper sheets separated with synthetic non-woven support fabric and placed on a grid were immersed for a given length of time at room temperature and under magnetic stirring in the treatment solution (1 L) containing the desired concentration of AAAS in HMDS (wt/wt). After treatment, the sheets were dried at room temperature under vacuum for one hour. Control papers were not subjected to any treatment as it was shown that the immersion in HMDS did not modify the mechanical properties of the paper.
Physico-chemical determinations
The alkaline reserve (AR) (meq(OH−)/100 g) was determined by back-titration according to the standard method ASTM D4988-96R01. The cold extract pH of the papers was determined according to TAPPI T509 om-88, with a 50 mg mass of paper. The AEAPMDMS uptake in the paper (% wt/wt) was measured by weighing the samples pre-conditioned at 23 °C and 50% relative humidity (rH) before and after treatment. The reported values are the average of the measurements on at least four sheets of paper.The tensile modulus at break (MPa), elongation at break (%) and tensile energy absorption (TEA) were measured from the stress–strain curves obtained with a tensile tester Adamel Lhomargy (DY-20B), according to the standard method NF: Q03-004 July 1986, where a coaxial stress is applied to the paper sample. Samples were tested at a speed of 10 mm min−1, with a 100 DaN load cell. The data was processed with TestWorks 4 (MTS Systems Corp.) software. The brittleness index (BI) was determined from the stress–strain curves according to Zou et al.17 The BI is proposed as the ratio of the elastic strain energy to the total TEA at the time of failure. A low value (BI ≪1) is characteristic of a ductile paper while a high value (BI ∼1) represents a brittle paper.
Zero-span tensile strength (zsTS) was measured with a Pulmac instrument (TS 100) according to TAPPI standard T231 cm-96. The value obtained (P) is transformed applying the formula zsTS = (P−P0) × 0.372 (Kgf 15 mm−1) modified to (P−P0) × 5.64 (daN mm−1), where P0 = 2 (constant of the instrument).
Folding endurance (FE) (the log of the number of double folds) was determined with a Tinius Olsen double fold instrument, according to ISO 5626![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1993. The applied force was 0.5 kg.
:1993. The applied force was 0.5 kg.
These mechanical properties were measured in the machine direction of the paper, on 10 strips taken from the same sample conditioned at 23 °C and 50% rH. Reported values are the average of the 10 measurements.
Color measurements were carried out with a hand-held spectrophotometer SP 64 (X-rite) equipped with an integrated sphere. The configuration adopted was in reflectance mode (spectral range 400–700 nm in 10 nm steps), with the specular component included, using the 5 mm diameter aperture. The colorimetric coordinate values (L, a, b)* were calculated in the CIE*Lab76 Color System, with a D65 standard illuminant and 10° standard observer.18 Based on the (L, a, b)* values measured before and after treatment and/or before and after artificial aging, the total color change (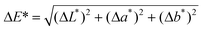 ) that occurred on a given sample was calculated.
) that occurred on a given sample was calculated.
A variable pressure scanning electron microscope JEOL JSM-5410LV was used to examine the samples. The accelerating voltage was 20 kV and the pressure was 0 Pa. Samples were observed using back-scattered electron imaging providing atomic number contrast to detect the location of specific atoms within the samples. The mappings were done over the same area by energy dispersive X-ray spectrometry (EDS) by means of an Oxford Instruments Link PentaFET Si(Li) detector and ISIS Link software. The EDS operating conditions were as follows: accelerating voltage 20 kV, ATM windows, take off angle 30°, working distance 20 mm, count time 50 s, approximate dead time 15%. The samples were gold coated before analysis.
Proton nuclear magnetic resonance (1H NMR) was carried out on a Bruker Avance 300 instrument working at 300 MHz with a frequency lock on deuterium. Liquid samples were introduced in 5 mm-diameter tubes. Spectra were scanned at room temperature. Solid-state {1H-29Si} cross polarization magic angle spinning NMR (CP MAS-NMR) was carried out on a Bruker ASX500 at 99.3 MHz (11.7 T). The 1H pulse width (π/2) was 6 μs, the contact time was 4 ms and the recycle delay was 20 s. The zirconia rotor of 4 mm-diameter was spun at 5 kHz. Chemical shifts were referred to an external reference, tetramethylsilane (TMS).
X-ray photoelectron spectroscopy (XPS) measurements were performed with a VG 220i-XL ESCALAB photoelectron spectrometer. A non-monochromatized Mg-Kα (1253.6 eV) source was used for X-rays. The acquisition of high-resolution spectra was made at a pass energy of 20 eV. A flood gun was activated for charge compensation. The binding energies were reliable to ±0.1 eV. Depth profile measurements were accomplished with an argon gun during a predetermined period of time and with an ion current that led to a sputter rate of approximately 0.2 nm s−1. The etch-acquired cycle was performed until further information from the system was obtained. The fitting of the XPS spectra was performed using Avantage software provided by ThermoFisher Scientific.
Results and discussion
Study of the reactivity of AEAPMDMS toward cellulose using 1H and 29Si CP MAS NMR spectroscopy
The reaction pathway of AEAPMDMS to form the corresponding di-aminoalkylalkoxysilanediol is shown in Fig. 1 (top). Fig. 1 (bottom left) shows the 1H NMR spectrum of a D2O extract of P2 paper treated with AEAPMDMS. It can be observed that the peaks of the methoxy groups (δ = 3 ppm) are totally absent from the spectrum while the CH2 multiplets of the propyl and ethyl moieties are clearly visible at δ 0.65, 1.65 and 2.9 ppm. This shows that, when extracted from the paper, AEAPMDMS is in the completely hydrolyzed form. The methanol produced by this reaction was lost during the drying under vacuum.In order to determine whether the hydrolyzed molecules of AEAPMDMS can condense in situ in the paper, solid state 29Si NMR spectroscopy with cross polarization was used on a sample of P2 paper treated with AEAPMDMS and defibrillated in a two-blade blender. The 29Si CP MAS-NMR spectrum is shown in Fig. 1 (bottom right). Two peaks are clearly visible at −15 and −22 ppm. According to the literature, the peak at −22 ppm corresponds to a D2 Si, i.e. a silicon atom linked to two –O–Si substituents.19,20 The peak at −15 ppm was assigned to a D1 Si, linked to only one –O–Si group, i.e. a terminal silicon atom. These results show that AEAPMDMS in the paper is polymerized. Although CP MAS-NMR is a semi-quantitative technique, the average number of monomers calculated from the ratio between the two peaks yields an approximate value of 10 as calculated from the relative intensity of the two peaks. These results are fully consistent with previous findings using AMDES, which undergoes the same hydrolysis and poly-condensation reactions in the paper fibers.6
Mass uptake of AEAPMDMS in the paper
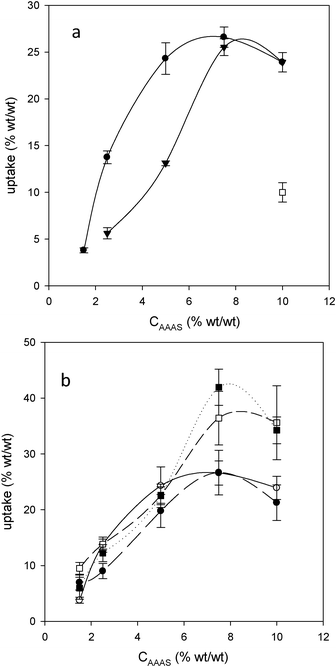 | ||
| Fig. 2 (a) Uptake for P2 as a function of treatment solution concentration, 15 min (▼) and 30 min (●) contact time in AEAPMDMS, and 30 min contact time (□) in AMDES.6 (b) Uptake as a function of AEAPMDMS concentration for P2 (○), P3 (●), B1 (■) and B2 (□) with 30 min contact time. | ||
SEM-EDS of B1 and B2
The diffusion of any active compound through the fiber walls is a parameter that is certainly worth considering for the efficiency of the deacidification and strengthening effect. In a previous study the even distribution and homogeneous penetration of several AAASs in the cellulose fibers was visualized using SEM-EDS.6 In that work, it was observed that silicon atoms were localized across the thickness of the paper sheet, with heavier deposits on the fibers' surface but also clearly, although to a somewhat lesser extent, throughout the interior of the fibers.In order to study the effect of the paper composition on the penetration of AEAPMDMS in the fibers, B1 (uptake 20%) and B2 (uptake 15%) were used. The cross sections of B1 show fibers in good morphological shape, most of them soft (chemical woodpulp), others with well-defined walls, hollow, thick, and wide open lumens (mechanical pulp), mostly from softwood (Fig. 3I). We also observe crevices, skirting dense areas. The EDS spectrum, not presented here, showed the elements Si and Al evidencing the presence of kaolin (Al2O3·2SiO2·2H2O). The paper was thus loaded with fillers in the pulp. After treatment with AEAPMDMS, the paper appears somewhat denser with fewer crevices. Moreover it underwent some morphological changes and became quite warped. The paper fibers appear slightly fused, but what stands out most is the seemingly “corrugated” aspect. The AEAPMDMS closed, merged and densified zones and somewhat plugged the holes. Transversely, the paper shows deep cracks, which probably reflect delamination. EDS mappings revealed the increase and the presence of Si and N in the fiber walls, indicating that the product has at least partly penetrated inside the fibers (Fig. 3I). A closer observation tends to show however that the lumens remain essentially devoid of AEAPMDMS, and that the highest density of Si and N is located in the external layers of the fiber walls.
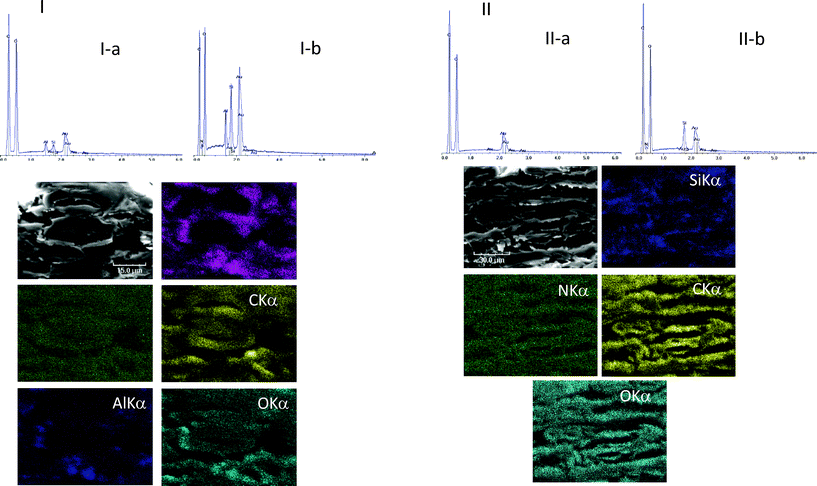 | ||
| Fig. 3 I(top): EDS spectra of the surface of B1 untreated (I-a) and treated with AEAPMDMS (I-b). I(bottom): SEM micrographs of a cross section of B1 treated with AEAPMDMS and EDS atomic mappings of Si, N, C, Al and O in corresponding areas. II(top): EDS spectra of the surface of B2 untreated (II-a) and treated with AEAPMDMS (II-b). II(bottom): SEM micrographs of a cross section of B2 treated with AEAPMDMS and EDS atomic mappings of Si, N, C and O in corresponding areas. | ||
In B2 the proportion of mechanical pulp is higher than in B1. Some fibers appear rigid, with open, and well distributed lumens. Unlike in B1, the fact that more entire and less dislocated fibers remain can be a sign of a disk refiner pulp (TMP = thermomechanical pulp or CTMP = chemi-thermomechanical pulp) rather than a groundwood pulp. Moreover, from the global aspect of the fibers and walls, it is likely that this paper may not contain chemical wood pulp. The EDS analysis, not shown here, confirmed the absence of mineral fillers, neither calcium or silicon nor aluminum were present.
After the treatment with AEAPMDMS, the sheets of B2 seem to have undergone less deformation than B1. Instead, the fibers’ structure appeared more closed, with less delamination. The result of impregnation with AEAPMDMS seems to be that of a more rigid paper. A narrowing of the lumens is also visible, as well as a narrowing of the spaces between the fibers. The EDS spectral analysis and elemental mappings (Fig. 3II) showed the presence of Si and N throughout the thickness of the paper indicating that the AAAS had penetrated into the fiber bed, and, at least partially, into the fiber wall.
XPS of P2 untreated and treated with AEAPMDMS
XPS is a surface analytical technique widely used for investigating the chemical composition of the near-surface of materials, with an analysis depth of up to 5 nm. Information at depths greater than 5 nm can be obtained by the ion etching of the sample and re-analysing. XPS provides quantitative chemical information as well as the oxidation and structural environments of all the elements, except hydrogen and helium. XPS proved to be a powerful technique for the chemical analysis and study of the elemental composition of a large variety of polysaccharide materials.21–23 It has also been used to examine the surfaces of paper that contain polymer additives24 and to specifically obtain chemical information on a wide variety of coated paper surfaces.25 However, little information is available on the use of XPS to study the penetration of dry strength agents in cellulose.In the first set of experiments, the untreated and AEAPMDMS treated papers were analyzed. Fig. 4A shows the XPS survey spectra of paper P2 before and after AAAS impregnation. For the untreated cellulose, carbon (at ∼285 eV) and oxygen (at ∼531 eV) were observed, with the experimental composition (C 59.9 at.%, O 40.1 at.%) close to the theoretical value for the cellulose repeat unit (C6H10O5). Not unexpectedly the O/C ratio decreased from 0.67 in the original sample to 0.25 in the AEAPMDMS treated sample, due to a lower oxygen atom concentration (Table 1). In contrast, the treated cellulose showed additional peaks at ∼399 eV corresponding to N1s, at ∼153 eV corresponding to Si2s and at ∼102 eV corresponding to Si2p.
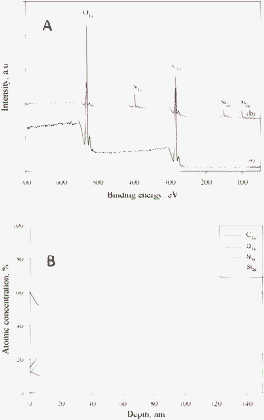 | ||
| Fig. 4 (A) XPS survey spectra of (a) untreated and (b) treated cellulose. (B) The depth concentration profile for the treated cellulose from paper P2. | ||
| Sample | C1s | O | N1s | Si2p | O/C ratio |
|---|---|---|---|---|---|
| Untreated cellulose | 59.9 | 40.1 | — | — | 0.67 |
| AEAPMDMS treated cellulose | 60.4 | 15.1 | 12.7 | 11.8 | 0.25 |
According to the literature, the assignment of the decomposed C1s peak for cellulose materials, which corresponds to the four types of carbon atoms, is well established (not shown).26 The peak at 284.3 eV corresponds to a carbon atom bound only to other carbon atoms and/or hydrogen atoms. The peak at 286.3 eV is due to a carbon bound to a single non-carbonyl oxygen atom. The peak at 288.2 eV represents a carbon atom bound to one carbonyl oxygen or two non-carbonyl oxygen atoms. The peak at 289.2 eV represents a carbon atom linked to one carbonyl oxygen and one non carbonyl oxygen. As one can see, the C1s spectrum of the treated cellulose was more complex (Fig. 5a). In addition to C–C, O–C–O and C–O peaks from the cellulose, new peaks appeared, which correspond to C–Si (283.3 eV) and C–N (285.2 eV and 287.3 eV) bonds. The fitted high resolution Si2p spectrum is shown in Fig. 5b. The binding energies of silicon chemical bonds can be located at 101.4 eV for R3–SiO and at 102.0 eV for C2–SiO2.26 It is established that the R3–SiO chemical bond has the highest atomic concentration. A decrease in the oxygen content (15.09 at.% for treated cellulose in comparison with 40.1 at.% for untreated cellulose) was revealed (Table 1). Also, the O/C ratio for treated cellulose was found to be equal to 0.25 (Table 1). This result, as well as the decomposition of the C1s spectrum (Fig. 5a), can be explained by the presence of AEAPMDMS in the paper.
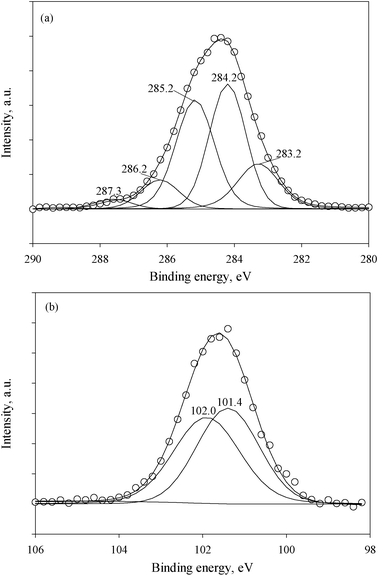 | ||
| Fig. 5 The fitted high resolution C1s (a) and Si2p (b) XPS spectra of cellulose after treatment with AEAPMDMS. | ||
To better understand the interaction of AEAPMDMS and cellulose, and to investigate the treatment effect on the depth penetration of the AEAPMDMS in the cellulose fibers, XPS sputtering was carried out. From the depth concentration profile obtained (Fig. 4B), the presence of Si and N was observed throughout depths of more than 140 nm, which testifies to the penetration of AAAS inside the cellulose fibers’ structure (the diameter size range of cellulose microfibrils being 2 to 30 nm, depending on the plant origin).27
Physico-chemical determinations and mechanical testing
:1994).28 The uptakes were quite similar for P2, P3 and B1 (around 6%), while it was over 9% for B2. The values for the uptakes did not exactly follow the same trend as the values for the alkaline reserve. A similar uptake for P2 and P3 led to a large difference in their respective ARs, while a large difference in the uptake for B1 and B2 led to a proportionally much smaller difference in their respective ARs. In other words, it seems that the papers containing no lignin and low amounts thereof (P2 and B1) show comparatively larger ARs in relation to their uptake than those with larger amounts of lignin (P3 and B2). This is consistent with the observation made above that, due to its carboxylic content, lignin ‘buffers’ part of the alkalinity initially introduced.
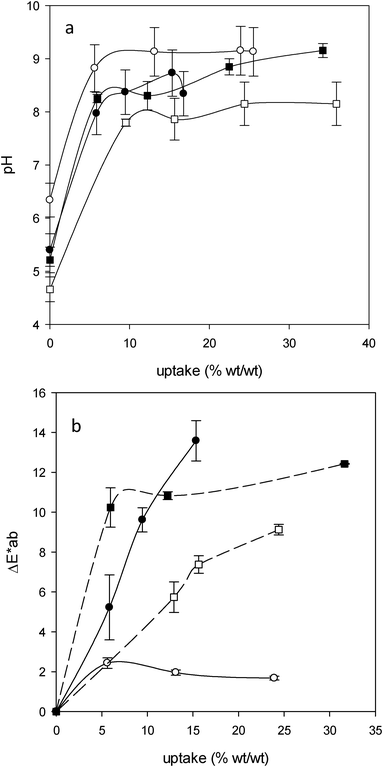 | ||
| Fig. 6 (a) Cold extract pH versus AEAPMDMS uptake for P2 (○), P3 (●), B1 (■) and B2 (□). (b) ΔE*ab as a function of AEAPMDMS uptake for P2 (○), P3 (●), B1 (■) and B2 (□). | ||
| Uptake % (wt/wt) | AR (meqOH−/100 g) | |
|---|---|---|
| P2 | 5.6 ± 1.2 | 28.0 ± 1.0 |
| P3 | 5.8 ± 0.9 | 21.4 ± 3.1 |
| B1 | 6.0 ± 1.9 | 35.7 ± 4.6 |
| B2 | 9.4 ± 1.5 | 39.0 ± 3.5 |
Mechanical properties
In Fig. 7c the zero span tensile strength (zsTS) of the papers as a function of the AEAPMDMS uptake is represented. A significant improvement in fiber strength was observed for P2 and P3 even for uptakes as low as 5%–6%. As the zsTS measures mostly the resistance of the fiber walls, it appears that the addition of even small amounts of AEAPMDMS strengthens the latter and to a lesser extent the bonds between the fibers. For comparison, the improvement in P2 was comparable to the strengthening achieved with AMDES at uptakes of 10% for Souguir et al.6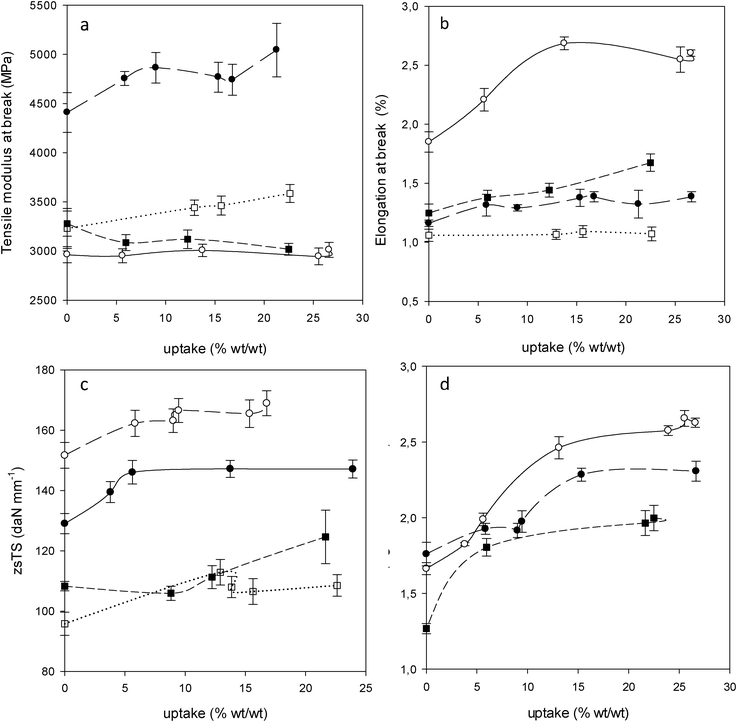 | ||
| Fig. 7 Mechanical properties as a function of AEAPMDMS uptake for P2 (○), P3 (●), B1 (■) and B2 (□). Tensile modulus at break (a), elongation at break (b), zero span tensile strength (zsTS) (c) and folding endurance (d) (0 double folds for B2). | ||
This is a logical consequence of the penetration of AAAS into the fibers as observed earlier with SEM and XPS. It can also be noted that the values of zsTS reach a plateau already at relatively low uptakes. This can be explained by a rapid saturation of AAAS in the fiber walls. For B1 and B2, an improvement in the fiber strength was observed but in a more moderate fashion in comparison with P2 and P3. This is probably due to the advanced state of degradation of the fibers of the books or a smaller penetration of AAAS into the fiber walls.
Fig. 7a shows the tensile modulus at break as a function of the uptake for the four papers studied. For P2 and B1, the modulus did not vary significantly upon incorporation of AEAPMDMS. For P3 and B2, a slight increase in the modulus is observed after treatment. In contrast, for the elongation at break (Fig. 7b) a significant improvement of the deformation capacity was observed for P2 after treatment and a slight improvement was observed for B1. For P3, a small increase in the elongation at break was observed while for B2 the incorporation of AEAPMDMS did not modify the deformation capacity. These different behaviors may have a relation with the pulp composition as well as with the brittleness of the papers. The papers containing no lignin (P2) or a larger percentage of cellulose chemical wood pulp (B1) showed a better deformability improvement than those containing larger amounts of lignin, which showed small or no improvement (P3 and B2, respectively, the latter containing the most lignin). The brittleness index (BI) before and after treatment with AEAPMDMS 1.5% was measured (Table 3). The BI does not represent either the stiffness or the toughness of the paper but it can demonstrate the ability of a paper to deform and be ductile and/or fragile. From Table 3 it seems that the papers can be classified according to their decreased ductility as follows: P2, B1, P3 and B2. Here again, it appears that the presence of a high percentage of lignin in the paper decreases its ductility. This ranking remains the same after treatment, but there was a slight decline in the BI for all the papers, which implies an improvement of the deformation capacity, especially so for P2 which, with the largest BI decrease (35%), benefits highly from the treatment.
| P2 | P3 | B1 | B2 | |
|---|---|---|---|---|
| Untreated cellulose | 0.20 | 0.50 | 0.38 | 0.70 |
| AEAPMDMS treated cellulose | 0.13 | 0.45 | 0.37 | 0.68 |
The results of the tensile tests and measurements of the brittleness index of the papers were confirmed by the measurements of the double fold endurance (FE). For B2, which is characterized by the highest BI as well as the highest lignin content, the measurement of the FE was simply not possible (zero double folds), and besides, the treatment failed at improving this property. This can be explained by the stiffness and weakness of the paper. In contrast, for P2, P3 and B1, which all show BI ≤ 0.5, as well as measurable initial double fold endurance, the treatment with AEAPMDMS improved the FE. This result tends to indicate that the improvement of the FE strongly depends on the initial strength of the paper (Fig. 7d). A minimal initial measurable strength seems to be required, which from the present results would stand somewhere near 10 measurable double folds. Upon this prerequisite, AEAPMDMS can act as a plasticizer.
Therefore, one persistent downside which the use of AEAPMDMS has not solved is the case of the papers in an ultimate degradation state with no residual folding endurance, i.e. highly oxidized papers. Their treatment could not restore the folding endurance. Despite the introduction of the active compound, highly weakened fibers cannot withstand the applied stress. This is due to the complete loss of the amorphous regions of cellulose, leaving the crystallites more isolated in the microfibrils.
Colorimetric measurements
The colorimetric measurements showed that the treatment resulted in an increased yellowing of P3, B1 and B2 (Fig. 6b). This phenomenon had been observed previously with AMDES.6 In contrast, P2 did not display a yellowing level that could be distinguished by the average observer at any uptake (ΔE* ≤ 2). As explained before, from a chemical standpoint the difference between P2 and the other papers is mainly the presence of various amounts of lignin (and additives) and the degree of oxidation of cellulose (B1 and B2). It has been proposed that the presence of carbonyl functional groups in the cellulose and in the other paper components, especially lignin, could lead to reactions with the primary amines of AAAS to form imine functions and other Schiff bases that yield color to the treated paper.29,30 This is, of course, a problem which can be considered as relative, depending on the initial state of yellowing and on the type of information carried by the paper.Conclusion
Significant uptakes of AEAPMDMS in papers were achieved, the extent of which depended largely on the pulp composition, with lignin playing a major role. The hypothesis for the approach of AAAS towards cellulose being related to the amine functionalities via hydrogen bonding was confirmed. Complete hydrolysis and poly-condensation of AEAPMDMS in situ was demonstrated and the additive was shown to deposit on the surface and inside the fibers, although lignin limited in-depth penetration. Despite some yellowing due to the likely formation of imines, improvement of the deformation capacity and mechanical strength of the paper through inter- and intra-fiber bonding was shown.Acknowledgements
A research grant from the French Ministry of Culture is gratefully acknowledged. Sabrina Paris from CRCC and Christine Labrugère from Centre de Caractérisation des Matériaux Avancés University Bordeaux 1 (France) for XPS investigations and fruitful discussions are warmly thanked.References
- H. A. Carter, J. Chem. Educ., 1996, 73, 417–420 CrossRef CAS.
- K. Turko, MMS Deacidification Systems: Planning and Managerial Decision Making, In Association of Research Libraries, Washington D.C., 1990, p. 33 Search PubMed.
- S. Ipert, E. Rousset and H. Cheradame, Restaurator, 2005, 26, 250–264 CrossRef CAS.
- S. Ipert, A.-L. Dupont, B. Lavédrine, P. Bégin, E. Rousset and H. Cheradame, Polym. Degrad. Stab., 2006, 91, 3448–3455 CrossRef CAS.
- A.-L. Dupont, B. Lavédrine and H. Cheradame, Polym. Degrad. Stab., 2010, 95, 2300–2308 CrossRef CAS.
- Z. Souguir, A.-L. Dupont, J.-B. d'Espinose de Lacaillerie, B. Lavédrine and H. Cheradame, Biomacromolecules, 2011, 12, 2082–2091 CrossRef CAS.
- M. Jacob, K. T. Varughese and S. Thomas, Biomacromolecules, 2005, 6, 2969–2979 CrossRef CAS.
- D. Pasqui, A. Atrei and R. Barbucci, Biomacromolecules, 2007, 8, 3531–3539 CrossRef CAS.
- S. H. North, E. H. Lock, C. J. Cooper, J. B. Franek, C. R. Taitt and S. G. Walton, ACS Appl. Mater. Interfaces, 2010, 2, 2884–2891 CAS.
- J. H. Moon, J. W. Shin, S. Y. Kim and J. W. Park, Langmuir, 1996, 12, 4621–4624 CrossRef CAS.
- A. K. Bledzki and J. Gassan, Prog. Polym. Sci., 1999, 24, 221–274 CrossRef CAS.
- M. N. Belgacem and A. Gandini, Compos. Interfaces, 2005, 12, 41–75 CrossRef CAS.
- M. Abdelmouleh, S. Boufi, M. N. Belgacem, A. P. Duarte, A. B. Salah and A. Gandini, Int. J. Adhes. Adhes., 2004, 24, 43–54 CrossRef CAS.
- M. S. Rakotonirainy, A.-L. Dupont, B. Lavédrine, S. Ipert and H. Cheradame, J. Cult. Heritage, 2008, 9, 54–59 CrossRef.
- V. Bennevault-Celton, O. Maciejak, B. Desmazieéres and H. Cheradame, Polym. Int., 2010, 59, 1273–1281 CrossRef CAS.
- STEP project CT 90 90-0100, TNO report BU3.94/1068/JH, 1994.
- X. Zou, N. Gurnagul, T. Uesaka and J. Bouchard, Polym. Degrad. Stab., 1994, 43, 393–402 CrossRef CAS.
- R. T. Marcus, AZimuth, 1998, 1, 31–96 CrossRef.
- Y. El Kortobi, J.-B. d'Espinose de la Caillerie, A.-P. Legrand, X. Armand, N. Herlin and M. Cauchetier, Chem. Mater., 1997, 9, 632–639 CrossRef CAS.
- T. -M. Lee, C.-C. M. Ma, C. W. Hsu and H. L. Wu, Polymer, 2005, 46, 8286–8296 CrossRef CAS.
- J. Li, Y. Wan, L. Li, H. Liang and J. Wang, Mater. Sci. Eng., C, 2009, 29, 1635–1642 CrossRef CAS.
- R. A. N. Pertile, F. K Andrade, C. Jr. Alves and M. Gama, Carbohydr. Polym., 2010, 82, 692–698 CrossRef CAS.
- M. Dobromir, G. Biliuta, D. Luca, M. Aflori, V. Harabagiu and S. Coseri, Col. Surf. A: Physicochem. Eng. Asp., 2011, 381, 106–110 CrossRef CAS.
- S. Chen and H. Tanaka, J. Wood Sci., 1998, 44, 303–309 CrossRef CAS.
- G. Strom and G. Carlsson, Nord. Pulp Pap. Res. J., 1993, 8, 105–112 Search PubMed.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers, Wiley, Chichester, UK, 1992 Search PubMed.
- M. Fujita and H. Harada, in Ultrastructure and Formation of Wood Cell Wall, Wood and cellulosic chemistry, ed. David N. -S. Hon and N. Shiraishi, Marcel Dekker Inc., 2nd edn, 2001, pp. 1–49 Search PubMed.
- International Standardization ISO 9706:1994. Information and documentation—paper for documents—requirements for permanence, International Organization for Standardization, Geneva, Switzerland.
- J. Martínez Urreaga and M. U. De la Orden, Eur. Polym. J., 2006, 42, 2606–2616 CrossRef.
- M. U. De la Orden and J. Martínez Urreaga, Polym. Degrad. Stab., 2006, 91, 886–893 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |