A close-up on doxorubicin binding to γ-cyclodextrin: an elucidating spectroscopic, photophysical and conformational study†
Resmi
Anand
,
Stefano
Ottani
*,
Francesco
Manoli
,
Ilse
Manet
and
Sandra
Monti
*
Istituto per la Sintesi Organica e la Fotoreattività, ISOF-CNR, via P. Gobetti 101, 40129, Bologna, Italy. E-mail: monti@isof.cnr.it
First published on 30th January 2012
Abstract
The association of doxorubicin (DOX) with γ-cyclodextrin (γ-CyD) was studied in phosphate buffer of pH 7.4, at 22 °C by performing titration experiments monitored with circular dichroism (CD), UV-vis absorption, and fluorescence. Global analysis of multiwavelength spectroscopic data obtained at different DOX concentrations was performed by taking into account the DOX monomer–dimer equilibrium. Formation of γ-CyD:DOX 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, 1:2 and 2:2 complexes was evidenced. The stability constants and the absolute CD, UV-vis absorption and fluorescence spectra of all the complexes were determined. γ-CyD did not prove to be able to disrupt the DOX dimer when the latter is the predominant form in solution. The triplet state absorption and kinetic properties of DOX in the presence of γ-CyD and in ethanol were also determined by laser flash photolysis. The excited singlet and the triplet features indicated the environment experienced by DOX in the CyD complexes is ethanol-like. The structure of the γ-CyD:DOX 1:1 complex was investigated by Molecular Mechanics (MM) and Molecular Dynamics (MD) for both the neutral and the positively charged (–NH3+ in the daunosamine moiety) DOX forms. The possibility of interaction of both the aglycone and the daunosamine parts with the γ-CyD cavity was evidenced.
:1, 2:1, 1:2 and 2:2 complexes was evidenced. The stability constants and the absolute CD, UV-vis absorption and fluorescence spectra of all the complexes were determined. γ-CyD did not prove to be able to disrupt the DOX dimer when the latter is the predominant form in solution. The triplet state absorption and kinetic properties of DOX in the presence of γ-CyD and in ethanol were also determined by laser flash photolysis. The excited singlet and the triplet features indicated the environment experienced by DOX in the CyD complexes is ethanol-like. The structure of the γ-CyD:DOX 1:1 complex was investigated by Molecular Mechanics (MM) and Molecular Dynamics (MD) for both the neutral and the positively charged (–NH3+ in the daunosamine moiety) DOX forms. The possibility of interaction of both the aglycone and the daunosamine parts with the γ-CyD cavity was evidenced.
Introduction
Anthracyclines represent an extremely important class of anticancer drugs, ranking among the most potent ones ever developed. They are usually employed in the treatment of leukaemia and various solid tumors, in particular breast cancer and Kaposi's sarcoma. They exert their action by interfering with DNA cutting and reclosing in a three component complex with DNA and Topoisomerase, thereby providing a cytostatic effect.1,2 The first anthracyclines isolated from the bacterium Streptomyces peucetius in the 60s were doxorubicin (DOX, also known as Adriamycin, Scheme 1) and daunorubicin (also known as daunomycin). Because of their great efficacy these two derivatives have been continuously applied in clinical practice, despite the development of forms of resistance by cancer cells and serious side effects related to low cardiac tolerability and necrotic action at the injection site.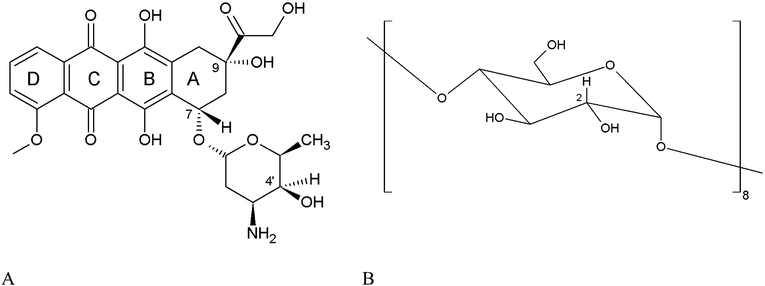 | ||
| Scheme 1 (A) Doxorubicin (DOX), (B) schematic of a γ-CyD glucopyranose unit. | ||
Looking for a better compromise between activity and toxicity of anthracyclines a lot of research efforts were invested in the identification of drug structural modifications. On the other hand, studies have focused on the development of improved delivery techniques based on the employment of biocompatible and biodegradable carriers, like micelles and liposomes,3 polymeric architectures,4–6 and nanoparticles.7–10 Delivery systems were also designed to contrast the tendency of these drugs to aggregate. Self-aggregation represents a serious drawback in their clinical use, because it may effectively compete with DNA binding thereby limiting the pharmacological activity. When incorporated in micelle-forming polymeric nanocarriers the DOX monomer displayed a major antitumor activity whereas the DOX dimer has no antitumor activity by itself.11
Cyclodextrins (CyDs) are biocompatible cyclic oligosaccharides, made of α-D-glucopyranose units joined by α(1–4) linkages (Scheme 1B), that form macrocycles with a hydrophilic exterior surface and a hydrophobic cavity able to host lipophilic guests. For a long time they have received considerable attention as carriers able to improve solubility, stability and bioavailability of drugs.12 Many derivatized CyDs and various CyD-based nanoassemblies, loading drugsvia weak non covalent interactions13–19 or via labile covalent bonds,6,20–22 have been synthesized and proposed as delivery systems in preclinical studies. The binding of DOX to CyDs has been addressed since the 90s. At that time it was reported that methyl-β-CyD potentiates the activity of DOX on both sensitive and multidrug-resistant cell lines.23–27 Since then, the interest for this topic has been continuously growing. Self-aggregation of DOX (evidenced in aqueous solution by UV-vis absorption,28 circular dichroism (CD)29 and NMR spectroscopy30) is likely perturbed by inclusion of the drug in a CyD cavity or a CyD nanoassembly. Therefore, gaining insights into the binding modes of DOX to CyD systems is of direct relevance to the optimization of the use of this drug.
As regards natural CyDs, DOX binds significantly to γ-CyD, whereas it possesses lower affinity for β-CyD and α-CyD.31–34 Formation of a γ-CyD-DOX complex with 1:1 stoichiometry has been reported.31,32,35 In this study we re-examined in detail the complexation of DOX with γ-CyD in aqueous medium, performing accurate titrations monitored with circular dichroism (CD), UV-vis absorption and fluorescence. Thanks to the different sign of the dichroic signal of monomer and dimer in the 500 nm absorption region, the CD technique yielded valuable information on the presence of the dimer and its disruption.36,37 By global analysis of multiwavelength CD data taken at different DOX concentrations we evidenced the formation of γ-CyD:DOX complexes of higher stoichiometries, i.e. 2:1, 1:2 and 2:2 in addition to 1:1, not reported previously, and we determined the stability constants and the individual CD features of all these complexes. We obtained information on the environment experienced by DOX in the CyD complexes from the excited singlet state properties of the fluorescent DOX monomer and from the triplet state properties of the non fluorescent DOX dimer by laser flash photolysis. We also studied the association of DOX with γ-CyD in the 1:1 stoichiometry from the structural point of view by Molecular Dynamics Simulations, with explicit solvent and examined the interaction of either the aglycone or the daunosamine moiety with the CyD cavity.
Materials and methods
Materials
Doxorubicin (DOX, Adriamycin), was purchased from ALEXIS Biochemicals and used without further purification; γ-CyD (Fluka) was used as received. Water was purified by passage through a Millipore MilliQ system. 0.01 M phosphate buffer at pH 7.4 was used. DOX was easily dissolved at a concentration of 2 × 10−4 M in this medium.Spectroscopic measurements
Ultraviolet absorption spectra were recorded on Perkin-Elmer Lambda 9 or Lambda 950 spectrophotometers. The spectra of the DOX-CyD mixtures were registered using CyD solutions as the reference. Circular dichroism spectra were obtained with a Jasco J-715 dichrograph. Fluorescence experiments were carried out on ∼1.0 × 10−5 M solutions in 1 cm cells having an absorbance of ≤0.1 at the excitation wavelength. Steady state fluorescence spectra were registered on a SPEX Fluorolog 111 spectrofluorimeter. Fluorescence lifetimes in air-saturated solutions were measured with a time correlated single photon counting system (IBH Consultants Ltd.). A nanosecond LED at 465 nm was used as the excitation source and the emission was detected at 590 nm. The software package for analysis of emission decays was provided by IBH Consultants Ltd. DOX fluorescence quantum yield was determined on excitation at 480 nm using Ru(bpy)3Cl2 as reference (Φ = 0.028, in air equilibrated solution).38CD and fluorescence titrations were recorded at constant drug concentration on varying CyD host concentration. The best complexation model and the association constants were determined by multivariate global analysis of multiwavelength data from a series of 9–11 spectra corresponding to different mixtures, using the program SPECFIT/32 (v.3.0.40, TgK Scientific). The optimization procedure is based on singular value decomposition and non linear regression modelling by the Levenberg–Marquardt method. The calculation also afforded the individual spectra of the associated species (see ESI† SI-4 for further details).
Nanosecond laser flash photolysis
The pulse of a Nd-YAG laser, operating at 532 or 266 nm (20 ns FWHM, 2 Hz), was suitably shaped passing through a rectangular, 3 mm high and 10 mm wide window, and providing a fairly uniform energy density, incident onto the sample cell (a pulse of 3.5 mJ at 266 nm corresponds to 12 mJ cm−2). A front portion of 2 mm of the excited solution was probed at a right angle, the useful optical path for analyzing light being 10 mm. A266 was ∼0.5–1 over 1 cm. Ar-saturated solutions were used. The sample was renewed after few laser shots. The temperature was 295 K.Computational methods
Interactions between γ-cyclodextrin (γ-CyD) and doxorubicin (DOX) was studied by Molecular Mechanics (MM) and Molecular Dynamics (MD) methods. The initial geometry of γ-CyD was taken from its crystallographic structure.39 For DOX, both the neutral and the positively charged molecules (–NH3+ in the daunosamine moiety) were considered. The initial atomic coordinates of these two species were obtained by ab initio quantum-mechanical geometry optimizations, using the Gaussian 09 program suite40 at the B3LYP/6-31+G(d,p) level of theory. Water solvent effects were simulated by the conductor-like polarizable continuum model (CPCM).According to the method proposed by Raffaini et al.,41 different initial molecular arrangements were prepared, as the DOX is alternatively rotated by 180°, around an axis perpendicular to the ring plane, to place the aglycone moiety facing the γ-CyD secondary (larger) rim (structure I) or far from it (structure II). Two additional arrangements have been prepared with the DOX molecule facing the γ-CyD primary (narrower) rim with the aglycone (structures Ib) and the daunosamine (structure IIb), respectively. These four geometries are reported in Scheme 2 and have been used as the starting geometries for both the neutral DOX and the positively charged DOX (–NH3+ in the daunosamine moiety). Initial geometries with the charged DOX molecules are labelled by adding the suffix plus to the previously defined labels. However, the charged structures are not reported in Scheme 2 since they differ only by a proton.
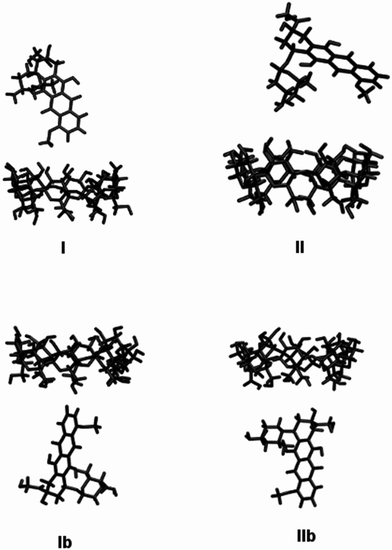 | ||
| Scheme 2 Initial geometries for γ-CyD/DOX system assumed for Molecular Mechanics (MM) and Molecular Dynamics (MD) calculations: I and II, aglycone and daunosamine units facing secondary γ-CyD rim, respectively; Ib and IIb, aglycone and daunosamine units facing the primary γ-CyD rim, respectively. | ||
Initial geometries, topologies and force field parameter files were prepared by the package Antechamber,42 using the General Amber Force Field (GAFF).43Antechamber was also used to solvate the molecules in a water box of appropriate size using the TIP3BOX water model. The periodic box used in MM and MD enclosed a total of ∼2050 water molecules. Moreover, the excess positive charge in the protonated systems was compensated by a Cl− ion. The MM and MD runs were performed by the package NAMD,44 using the compatibility features with the GAFF. Initial geometries were minimized and optimized and subsequently heated up to 300 K. The systems were allowed to relax and equilibrate at this temperature and at a pressure of 1 atm for a simulation time of 1 ns. MD production runs were carried on at constant temperature and pressure by using Langevin dynamics for an additional simulation time of 10 ns, at least. MD runs were performed with completely unconstrained bonds and a time step of 0.5 fs was used for compatibility with the vibrational frequencies of bonds with hydrogen atoms. Frames with the system geometries have been recorded at regular time intervals along these trajectories. The molecular geometries reported in this work were obtained by the graphical package VMD.45VMD plugins were used to perform some of the analysis of the MD trajectories.
Results and discussion
Selfassociation of DOX in buffer
The absorption spectrum of DOX in phosphate buffer at pH 7.4 displays bands at 288 and 480–500 nm relevant to the two allowed 1A → 1La and 1A → 1Lb π,π* transitions, polarized along the short and long axis, respectively.29 A shoulder around 320–380 nm is associated to n,π* transitions of the three C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the molecule, partially forbidden by electric dipole.36 Self-aggregation of DOX affects the band shapes and the molar absorption coefficients that tend to decrease at increasing concentrations. The spectral profile in the visible region is strongly influenced by the protonation state of the aglycone moiety, but is practically insensitive to protonation of the daunosamine moiety. At pH 7.4 the aglycone part is neutral, whereas the daunosamine is protonated.29,36 Bands at 252 and 233 nm have been assigned to the aglycone moiety,46 with some contribution of the daunosamine moiety.30
O groups in the molecule, partially forbidden by electric dipole.36 Self-aggregation of DOX affects the band shapes and the molar absorption coefficients that tend to decrease at increasing concentrations. The spectral profile in the visible region is strongly influenced by the protonation state of the aglycone moiety, but is practically insensitive to protonation of the daunosamine moiety. At pH 7.4 the aglycone part is neutral, whereas the daunosamine is protonated.29,36 Bands at 252 and 233 nm have been assigned to the aglycone moiety,46 with some contribution of the daunosamine moiety.30
At concentrations below 5 × 10−3 M a simple dimerization model is sufficient to describe DOX aggregation.30 A set of absorption spectra obtained upon DOX dilution in the range 5.0 × 10−5 M—1.0 × 10−7 M (see ESI,†SI-1, Fig. S1) was globally analysed adopting this model with the program SPECFIT/32 (see Materials and Methods and ESI,†SI-4). A dimerization constant with log(Kd/M−1) = 4.8 ± 0.1 was extracted, in reasonable agreement with the literature data.28,47,48 The spectrum of the DOX dimer in solution was also determined and is reported together with that of the monomer in the inset of Fig. S1 of the ESI.† It has been recently suggested by Agrawal et al. on the basis of 2D NOESY spectra that the geometrical arrangement of the two DOX units in the dimer consists of stacking of the aglycone moieties in either parallel or antiparallel orientation, with the methoxy substituent of the D ring pointing towards the exterior or the interior of the interplanar space, respectively (Scheme 3).30 The strong hypochromicity of the absorption bands of the DOX unit in the dimeric state (see ESI,† Fig. S1) is consistent with both arrangements.
 | ||
| Scheme 3 Proposed doxorubicin dimer structures, (A) parallel, (B) antiparallel arrangement.30 | ||
Anthracyclines are endowed with an intrinsic CD due to several asymmetric carbon centers. The C7 and C9 configuration are of particular importance in the explored wavelength region (Scheme 1).29,36 The CD spectrum of DOX 1.6 × 10−4 M is characterized by negative bands at 202 nm, 293 nm, 516–547 nm and positive bands at 233 nm, 252 nm, 352 nm and 453 nm. The positive–negative split dichroic signal in the 420–580 nm region is due to the presence of DOX in dimeric form. The presence of the amino sugar affects the Δε for the band corresponding to the π,π* transition polarized along the long axis, due to the enhancement of the DOX molecular dissymmetry.29 At DOX concentration ≤1.0 × 10−5 M the CD spectrum does not exhibit a prominent negative signal at 530–550 nm and suggests a predominance of the monomer over the dimer.
Association of DOX to γ-cyclodextrin
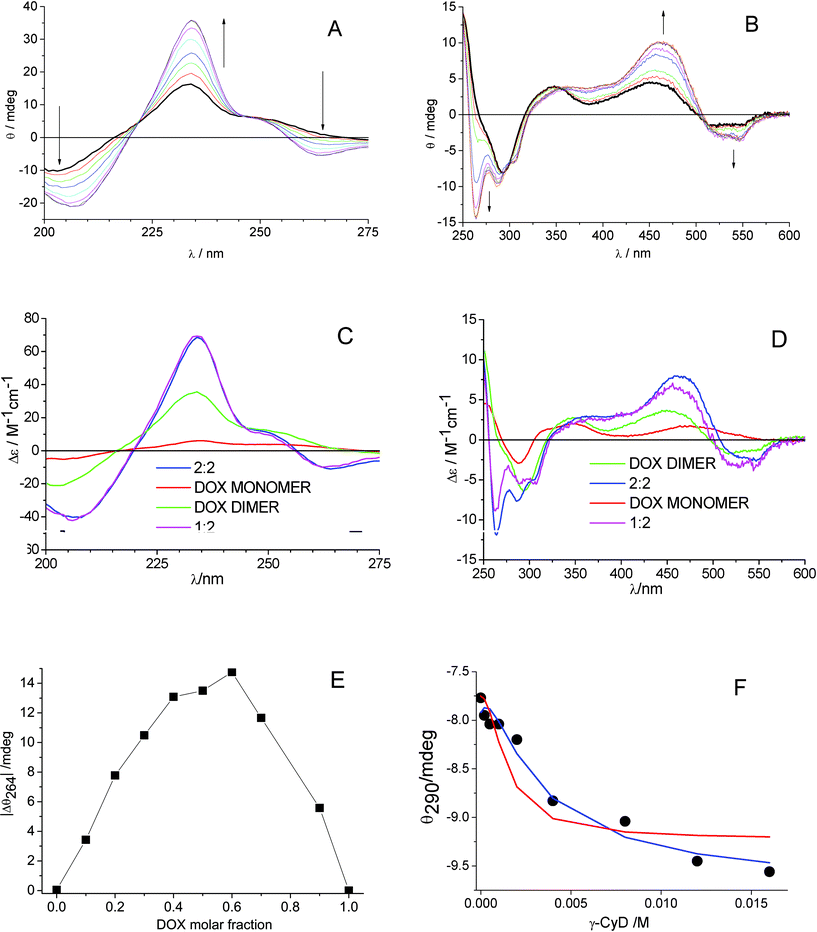 | ||
| Fig. 1 Ellipticity changes of DOX 1.6 × 10−4 M in 0.01 M phosphate buffer at pH 7.4 and 22 °C, titrated with γ-CyD in the concentration range 2.0 × 10−4 M - 1.6 × 10−2 M: (A) cell path 0.2 cm; (B) cell path 0.5 cm. The signal of γ-CyD alone was subtracted. (C), (D) Absolute CD spectra of DOX dimer (green), 1:2 γ-CyD:DOX complex (magenta) and 2:2 γ-CyD:DOX complex (blue) for log Kd/M−1 = 4.8 and log(K12/M−2) = 7.80 and log(K22/M−3) = 10.48. The spectrum of free DOX monomer (red) was fixed in the calculations. (E) Modified Job plot of absolute value of ellipticity of γ-CyD–DOX mixtures at 264 nm, (|Δθ264|), after subtraction of the signal of DOX alone, vs.DOX molar fraction, total concentration of DOX and γ-CyD = 10−3 M. (F) Comparison of experimental (•) and calculated (line) ellipticity at 290 nm for the model with 2:2 complex only (red, log(K22/M−3) = 10.8) and 1:2 + 2:2 complexes (blue). | ||
The sets of spectra of Fig. 1A and 1B were globally analyzed with SPECFIT/32. Several complexation models were tried, involving 1:1, 2:1, 1:2 and 2:2 γ-CyD:DOX complexes in various combinations. We included the DOX dimerization equilibrium with a fixed constant of log(Kd/M−1) = 4.8. The CD spectrum of DOX alone at concentration 5 × 10−6 M was measured in a 10 cm cell and was assigned to the DOX monomer and also fixed in the calculation. A good fit over the whole 200–600 nm range was found for a model with a single 2:2 CyD:DOX complex with association constant log(K22/M−3) = 10.8 ± 0.2, Durbin Watson (DW) factor of 1.5 (relative error of fit 3.1%) in the 200–280 nm range and DW =1.9 (relative error of fit 6.2%) in the 250–600 nm range. The individual spectra of all the species involved in the equilibria are shown in Fig. S2 of the ESI,†SI-2. Global analysis of the set of absorption spectra of Fig. S3 in SI-2 with the same model afforded log(K22/M−3) = 10.8 ± 0.1 (Durbin Watson factor 2.0) in excellent agreement with the CD result. The individual absolute absorption spectra of all the components are reported in the inset of Fig. S3 in the ESI,†SI-2. To confirm the stoichiometry of complexation a continuous variation experiment49 was performed at 1.0 × 10−3 M total γ-CyD + DOX concentration, a compromise between the drug limited solubility/aggregation tendency and the need to have distinctive signals from complexes in the 10−4–10−3 M γ-CyD concentration range. The absolute value of the ellipticity at 264 nm associated to the complexation progression (see Fig. 1B) was corrected subtracting the DOX intrinsic signal, (|Δθ264|) and was plotted vs. the DOX molar fraction (Fig. 1E). The plot is characterized by a broad asymmetric bell-shape profile with maximum at ca. 0.6 molar fraction. This indicates a significant, but not exclusive, presence of 1:2 complexes in the equilibrium mixture (exclusive presence of 1:2 stoichiometry would give a maximum at 0.7 molar fraction). Likely the contemporary presence of both the 1:2 and 2:2 stoichiometries is the best model to describe the γ-CyD complexation at “high” DOX concentrations. Indeed assuming this model analysis of the data of Fig. 1A and B afforded satisfactory fits with log(K12/M−2) = 7.80 ± 0.04 and log(K22/M−3) = 10.48 ± 0.21 (DW = 1.8 and relative error of fit 1.04% in the UV and DW = 2.4 and error of fit 5.42% in the UV-vis). The extracted spectra of the 1:2 and 2:2 complexes are represented in Fig. 1C,D. In the UV range the spectra appear almost identical to each other, both being characterized by negative peaks at 207 and 264 nm with Δε ≈ −40 and −10 M−1 cm−1, respectively, and an intense positive peak at 234 nm of Δε ≈ 70 M−1 cm−1. Also in the UV-vis range the CD spectra of the 1:2 and 2:2 complexes are similar to each other with positive/negative splitting of the lowest energy band and negative peaks of different relative size at 264, 288, 306 nm. In Fig. 1F the comparison between experimental and calculated ellipticity values is shown at 290 nm, a wavelength in a critical region, for the model with a 2:2 complex only and the model with both 1:2 and 2:2 complexes.
On the basis of the above analysis of the complexation equilibrium we can safely conclude that γ-CyD is not able to disrupt the DOX dimer when the latter is the predominant DOX form in solution, rather forming associates in 1:2 and 2:2 stoichiometry. Given the geometrical arrangement of the two DOX units in the dimer (Scheme 3),30 it is relevant to gain information about the possibility for either the aglycone or the daunosamine moieties to interact with the γ-CyD macrocycle. To this goal, in addition to a MM and MD investigation (see below), an acid–base titration monitoring the absorption of DOX in the pH 6–11 interval was carried out. Deprotonation of the daunosamine–NH3+ and of one of the phenolic OH groups of the aglycone ring B in aqueous medium occurs with pK values of ca. 8.15 and 10.16, respectively50,51 and produces large changes in both the UV region (252 and 233 nm bands) and in the visible region (Fig. 2A). Encapsulation of the sugar or the aglycone moieties in the CyD cavity was expected to perturb the relevant deprotonation process. Actually in the presence of γ-CyD 1.2 × 10−2 M (ca. 99% DOX complexed in either 1:2 or 2:2 stoichiometry with a large predominance of the latter one) the pH induced spectral modifications were much smaller over the whole 230–600 nm spectral range. This indicated higher pKs in the CyD environment for both the deprotonation steps and suggested the interaction of both the aglycone and the daunosamine moieties with the γ-CyD cavity.
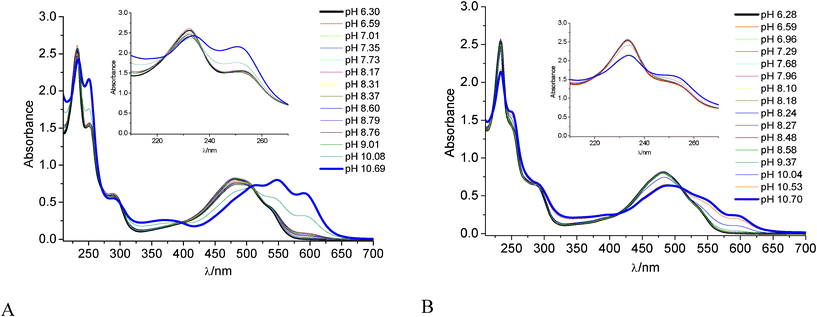 | ||
| Fig. 2 (A) Absorption spectra of DOX 1.7 × 10−4 M in 0.01 M phosphate buffer in the range of pH 6–11, reference water, 22 °C. (B) The same in the presence of γ-CyD 1.2 × 10−2 M; reference γ-CyD solutions. Cell 0.5 cm. Insets: detail of the 210–270 nm range. | ||
:1 in the absence of γ-CyD. Increasing γ-CyD concentrations induced intensity increase of both the positive band at 470 nm and the negative one at 288 nm; the signals at 252 nm, 233 nm and 202–205 nm also gained intensity but no new bands appeared in the visible. The sets of CD spectra in Fig. 3A and 3B were analysed again with various complexation models with 1:1, 2:1, 1:2 and 2:2 γ-CyD:DOX complexes in various combinations, all including the DOX monomer–dimer equilibrium. In no case the calculation attained convergence with inclusion of DOX monomer–dimer equilibrium, so we neglected it. The best model for the whole spectral window (200–600 nm) was that involving a contemporary presence of 1:1 and 2:1 γ-CyD:DOX complexes. The relevant optimized binding constants were log(K11/M−1) = 2.7 ± 0.2 and log(K21/M−2) = 4.4 ± 0.5, with DW parameters in the 1.7–2.4 range. The individual spectra of the various species are reported in Fig. 3C and 3D. The agreement between calculated and experimental ellipticities at representative wavelengths is shown in Fig. S6 of the ESI,†SI-3. We cannot exclude that omission of the dimerization equilibrium may have led to more approximate association constants and spectra for the complexes. However is worth noticing that the calculated binding constant for the 1:1 complex is in good agreement with values previously reported in literature34,35 and the extracted spectrum for the free DOX monomer is very close to that experimentally measured at 5 × 10−6 M (see Fig. 1). Global analysis of the UV-vis absorption titration data, performed upon fixing the 1:1 association constant, confirmed the magnitude of the 2:1 association constant (Table 1) and afforded the absolute absorption spectra of the 1:1 and 2:1 complexes (see Fig. S5 in the ESI,†SI-3).
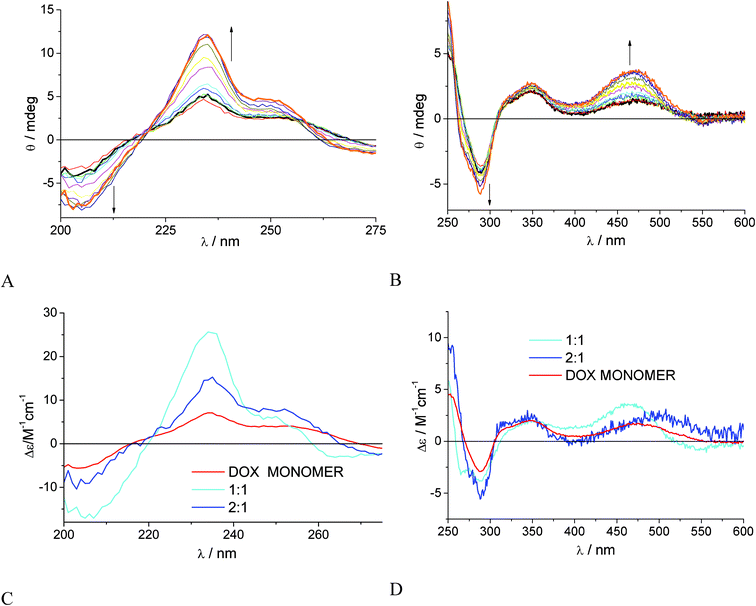 | ||
| Fig. 3 Ellipticity changes of DOX 1.0 × 10−5 M in 0.01 M phosphate buffer at pH 7.4 and 22 °C, titrated with γ-CyD in the concentration range 5.0 × 10−5 M–1.2 × 10−2 M: (A) cell path 2 cm; (B) cell path 4 cm. The signal of γ-CyD alone was subtracted. (C), (D) Absolute spectra of DOX monomer (red), 1:1 (cyano) and 2:1 (blue) γ-CyD:DOX complexes, for log(K11/M−1) = 2.7 ± 0.2 and log(K21/M−2) = 4.4 ± 0.5. | ||
| DOX complexes | Binding constants | Technique |
|---|---|---|
| DOX2 (DOX dimerization) | log(Kd/M−1) = 4.8 ± 0.1 | UV-vis absorption |
|
γ-CyD:DOX 2:2 |
log(K22/M−3) = 10.48 ± 0.21 | CD |
|
γ-CyD:DOX 1:2 |
log(K12/M−2) = 7.80 ± 0.04 | CD |
|
γ-CyD:DOX 1:1 |
log(K11/M−1) = 2.7 ± 0.2 | CD |
| log(K11/M−1) = 2.3 ± 0.3 | Fluorescence | |
|
γ-CyD:DOX 2:1 |
log(K21/M−2) = 4.4 ± 0.5 | CD |
| log(K21/M−2) = 4.9 ± 0.1 | Fluorescence | |
| log(K21/M−2) = 4.9 ± 0.4 | UV-vis absorption |
:1 and 2:1 complexes, log(K11/M−1) = 2.3 ± 0.3 and log(K21/M−2) = 4.9 ± 0.1 (Durbin Watson parameter 2.2). The individual fluorescence contribution of each species in solution is reported in Fig. 4B.
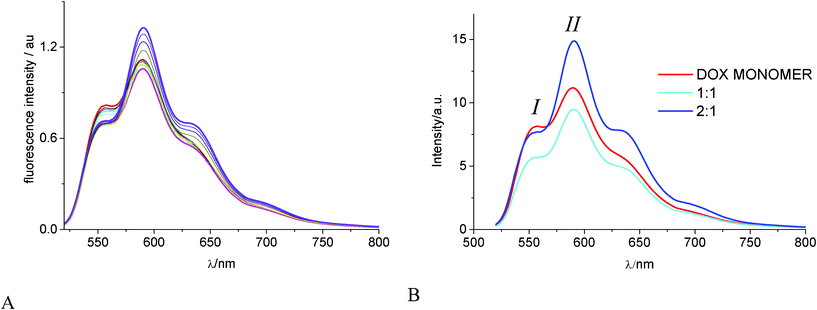 | ||
| Fig. 4 (A) Fluorescence intensity changes of DOX 1.0 × 10−5 M in 0.01 M phosphate buffer at pH 7.4 and 22 °C, upon titration with γ-CyD in the range 1.0 × 10−4 M–1.6 × 10−2 M. (B) Separated emission spectra of DOX monomer (red); 1:1 (cyano); 2:1 (blue) γ-CyD:DOX complexes, corresponding to log(K11/M−1) = 2.3 ± 0.3 and log(K21/M−2) = 4.9 ± 0.1 | ||
The area under each of the spectral profiles of Fig. 4B is proportional to the corresponding emission quantum yield (Φf). Using the value of Φf = 0.039 for DOX in buffer (see Materials and Methods), we obtained a value of Φf11= 0.032 for the 1:1 and Φf21= 0.048 for the 2:1 complex. The 550 nm/590 nm intensity ratio (I/II) in the emission spectrum of DOX is a parameter probing the environment of the hydroxyanthraquinone-centered emitting state, because from a value of ca. 0.80 for water, the ratio progressively diminishes in protic solvents of decreasing polarity (e.g. 0.57 in ethanol, 0.51 in 1-heptanol).52 In the spectra of Fig. 4BI/II changes from 0.73 in pure buffer to 0.60 in the 1:1 complex and 0.51 in the 2:1 complex. Thus the I/II values in the complexes tend to those of DOX in alcoholic solvents, indicating the excited state of the drug experiences close proximity with the hydroxyl groups of the CyD rim and feels a decrease in the environmental polarity.
The DOX fluorescence decay was little affected by the presence of γ-CyD. The decay kinetics was monoexponential with τf = 1.02 ns in buffer and 1.13 ns in presence of γ-CyD 5.0 × 10−3 M (DOX ≥ 80% complexed in 1:1 or 2:1 γ-CyD:DOX stoichiometries).53 Considering the relation of Φf and τf with the radiative (kr) and non radiative (knr) rate constants of the emitting state (eqn (1)):
| Φf = krτf = kr /(kr + knr) | (1) |
Triplet state of CyD:DOX complexes
The triplet state properties of the guest are generally useful to gain information on drug environment in CyD complexes.54Flash photolysis of a 1.6 × 10−4 M DOX solution in Ar-saturated phosphate buffer of pH 7.4 in the presence of 1.6 × 10−2 M γ-CyD was therefore carried out with 532 nm laser excitation. In these conditions > 96% of DOX is associated in 1:2 or 2:2 stoichiometry. A weak differential absorption with bands at 350, 420 and 480 nm was observed (Fig. 5A). Its decay appeared as monoexponential with time constant τ = 4.1 ± 0.1 μs. It was assigned to the population of the DOX triplet state.55 Given the adopted experimental conditions the actual assignment was to the triplet of the DOX dimer in the γ-CyD complex(es). Unfortunately, detection of the triplet absorption at DOX concentrations ≈ 1 × 10−5 M was not possible in our laser flash photolysis apparatus. Thus we could not directly reveal the triplet features of monomeric DOX in the γ-CyD complexes. However the spectral profile and the lifetime of the transient in Fig. 5A are similar to those observed in EtOH (see Fig. 5B), where the drug is monomeric. It can be inferred that the triplet state absorption is not significantly affected by the DOX pairing in the dimer (at least in its qualitative features) and probes an alcoholic environment, as expected on the basis of the excited singlet properties of the monomeric DOX complexes. It is worth observing the triplet lifetime in water is sensibly shorter (τ = 1.7 μs) than that in EtOH and in γ-CyD, see also Table 2.55
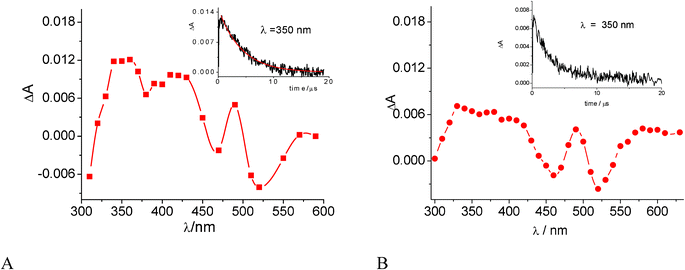 | ||
| Fig. 5 Difference absorption spectra observed 300 ns after excitation with a 20 ns laser pulse, cell path 1 cm, at 22 °C: (A) 1.6 × 10−4 M DOX in Ar-saturated 0.01 M phosphate buffer at pH 7.4 in the presence of 1.6 × 10−2 M γ-CyD, laser pulse 532 nm, 3 mJ. (B) 7.0 × 10−5 M DOX in EtOH, laser pulse 266 nm, 2 mJ. Insets: decay profiles at 350 nm. | ||
Analysis of molecular dynamics trajectories for γ-CyD:DOX 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 association
:1 association
The interaction between γ-CyD and DOX was studied by MM and MD modelling (see all the details of the computational methods in the Materials and Methods section). The main purpose of this study was to investigate the ability of the DOX molecule to interact with the γ-CyD cavity with different molecular portions, (A, B, C, D)-rings and the sugar moiety. Some tentative MD runs showed that, in a water box of appropriate size, the γ-CyD-DOX complex forms rather rapidly and, under constant pressure and temperature, the reciprocal arrangements of the partners are quite persistent in their main geometrical parameters, even for long MD runs. Table 3 reports some relevant data obtained from the analysis of the MD trajectories. All the averages in this table are computed by the values obtained at fixed time intervals along the trajectories. Average values of the total energies are all negative and their standard deviations are less than 1%. The mass-weighted radius of gyration, rgyr, has been obtained for the γ-CyD, the DOX and the complex by eqn (2):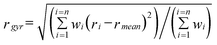 | (2) |
| Initial geometry | Energy average [kcal mol−1] | Interaction energy Average [kcal mol−1] | r gyr (complex) Average [Å] | r gyr (γ-CyD) Average [Å] | r gyr (DOX) Average [Å] |
|---|---|---|---|---|---|
| I | −16347 ± 114 |
−38 ± 5 | 6.17 ± 0.08 | 6.00 ± 0.08 | 4.75 ± 0.08 |
| II | −15129 ± 110 |
−41 ± 6 | 6.26 ± 0.08 | 6.55 ± 0.22 | 4.36 ± 0.07 |
| I+ | −17365 ± 118 |
−46 ± 7 | 6.12 ± 0.08 | 6.00 ± 0.07 | 4.77 ± 0.07 |
| II+ | −15623 ± 111 |
−31 ± 5 | 6.44 ± 0.17 | 6.24 ± 0.13 | 4.56 ± 0.06 |
| Ib | −17586 ± 120 |
−28 ± 3 | 6.32 ± 0.14 | 5.93 ± 0.20 | 4.78 ± 0.06 |
| IIb | −17671 ± 120 |
−28 ± 3 | 6.37 ± 0.10 | 6.09 ± 0.09 | 4.78 ± 0.10 |
| Ib+ | −19962 ± 122 |
−47 ± 3 | 6.14 ± 0.05 | 6.51 ± 0.09 | 4.82 ± 0.04 |
| IIb+ | −17787 ± 118 |
−22 ± 4 | 6.80 ± 0.17 | 6.08 ± 0.07 | 4.77 ± 0.07 |
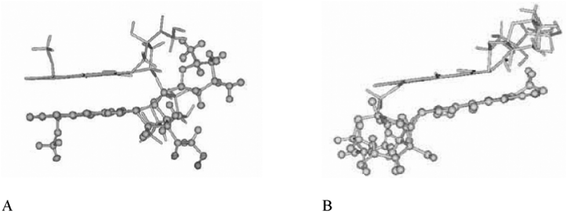
The stability of the complexes along the trajectories is consistently confirmed by their negative interaction energies and by the small standard deviations in the values of energy and radius of gyration. Fig. 6 reports some relevant structures extracted from the MD trajectories. The labels correspond to the initial geometries of the complexes in Scheme 2. As far as simulation conditions are concerned, these geometries should be regarded as illustrative of stable structures, since temperature fluctuations and collisions with solvent molecules are not able to separate molecules and convert to different complexes. Comparison of geometries in Fig. 6 with data in Table 3 shows that the Ib+ initial setting leads to the more stable complex, as confirmed by the lowest values of the Energy, Interaction Energy and rgyr. This final geometry corresponds to the one reported in the literature32 and it is achieved quite early (< 1 ns) in the MD trajectory, by insertion of the D-ring into the γ-CyD cavity through the primary (narrow) rim. Table 3 shows that interaction of DOX with the γ-CyD primary rim is energetically more favored, probably by stabilization with the solvent molecules since the interaction energies are not consistently lower as compared to complexes interacting on the secondary (large) rim. Actually, rgyr of the I and I+ complexes show that a good packing can be achieved even by interaction on the secondary rim and that especially the I+ structure can be of significance in the complex formation. Inspection of Fig. 6 suggests that hydroxyl groups on the γ-CyD rims interact preferably with the conjugated ring system of the DOX, particularly with rings B and C. This is confirmed by comparing the distribution of distances between the γ-CyD center of mass and the B or D-ring, respectively (see SI-5 Fig. S7). The B-ring is consistently closer to the γ-CyD center of mass than the peripheral D-ring, a trend which is even more pronounced in the Ib+ complex.
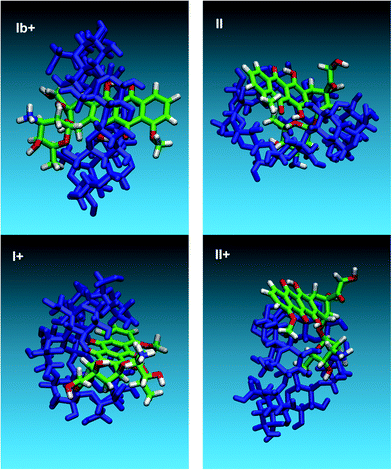 | ||
| Fig. 6 Typical geometric arrangements obtained in the MD simulations. The labels correspond to the initial settings in Scheme 2. | ||
Fig. 6 shows that insertion of the DOX daunosamine moiety into the γ-CyD cavity gives rise to stable complexes too. The –NH2group can cross from the secondary to the primary rim in complex II, while the positively charged –NH3+ is confined outside the secondary rim in complex II+. NOESY experiments performed by Bekers et al.32 showed that the distance between hydrogen atoms H2 of the γ-CyD and H4′ of the daunosamine is about 3 Å for a stable complex. However, the same investigators couldn't obtain optimized molecular models consistent with this result. This point has been investigated in the present work by computing the radial pair distribution function, g(r), for all possible pairs of the DOX H4' atom with the H2 atoms of the γ-CyD along the trajectories of the eight studied complexes. The pair distribution function is defined as the probability of finding a second particle as a function of distance from an initial particle and its values for the investigated complexes are reported in Fig. 7. Data in this figure show that only complex II+, with the daunosamine moiety into the γ-CyD cavity, comes close to the NOESY experimental constraint. All other complexes, even the most stable Ib+, display probabilities that become significant only at larger distances. Results in Fig. 7 are also consistent with the distance of 5.7 Å for the neutral DOX-(S)-isomer obtained previously by molecular modeling.32 According to the geometries in Fig. 6 and data in Fig. 7, it can be concluded that a distance of 3 Å is not compatible with the daunosamine H4' atom interacting with the exterior of the γ-CyD molecule,32 which would correspond to larger distances. Instead, it should stem from contributions of structures similar to complex II+, where the daunosamine moiety is inside the γ-CyD cavity. The structure of these complexes suggests a possible rationale for the interaction of the DOX molecule with two γ-CyD units (2:1 complex) as well as for the interaction of the DOX dimer with two γ-CyD units (2:2 complex). Higher order complexes will be the object of further molecular modelling investigations.
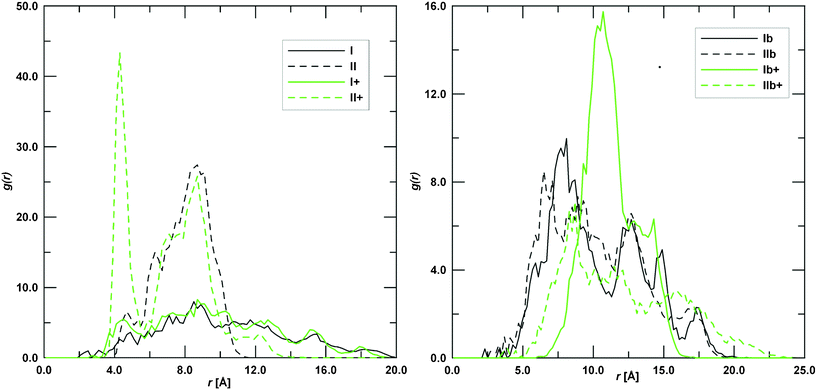 | ||
| Fig. 7 Radial pair distribution function, g(r), of the possible pairs between the DOX H4' atom with the H2 atoms of the γ-CyD. Values corresponding to interactions with the secondary and the primary rim of the γ-CyD are reported in the left and right plot, respectively. | ||
Concluding remarks
The γ-CyD-DOX complexation in aqueous buffer is a process characterized by a great intrinsic complexity, mainly due to the DOX self-aggregation. In the present study the complexation equilibria were analysed taking into account the DOX monomer–dimer equilibrium, but it was not considered that the DOX dimer exists in two conformations, parallel and antiparallel, each of them expected to have a peculiar affinity for γ-CyD. So, our analysis yields a single spectrum for a given stoichiometry even though it may represent different conformations. In spite of this approximation, UV-vis absorption, CD and fluorescence clearly proved the formation of multiple γ-CyD:DOX complexes. 1:1, 2:1, 1:2 and 2:2 association stoichiometries were evidenced for γ-CyD ≤ 1.6 × 10−2 M. The accurate study of the CD allowed to demonstrate that the complexes of monomeric DOX with one (1:1) or two CyD units (2:1) prevail at DOX concentrations ≤10−5 M, whereas complexes of dimeric DOX with one (1:2) or two γ-CyD units (2:2) dominate at DOX concentrations > 10−4 M. The formation of higher order γ-CyD:DOX complexes was not reported in previous literature.32 The stability constants of all the complexes, determined by global analysis of titration data from several spectroscopic techniques, showed good self-consistency (Table 1), which reinforces the reliability of the individual CD, fluorescence and UV-Vis absorption spectra for a given stoichiometry (Fig. 1, 3, 4 and Fig. S1, S4 and S5 in the ESI†).
The CD spectra of the 1:1 and 2:1 complexes in the UV region, when compared to each other, point to opposite dichroic contributions for the association of the first and the second γ-CyD unit to monomeric DOX (Fig. 3C); the corresponding profiles in the visible (Fig. 3D) indicate the CD is increased in intensity and blue shifted in the 1:1 complex and somewhat decreased in intensity and red-shifted in the 2:1 complex. These features suggest the aglycone moiety interacts with γ-CyD in the first complexation step while the daunosamine unit interacts in the second one.56
The MM and MD trajectories support the CD interpretation. Geometries in Fig. 6 (Ib+) and data in Table 3 show that the most favourable interaction is that with DOX aglycone part approaching the γ-CyD primary rim. The complex geometry is characterized by the hydroxy-anthraquinone core (ring C and B) embedded in the cavity with long axis parallel to the γ-CyD axis and ring D protruding out of the CyD secondary rim. A further possible complex geometry is that represented in Fig. 6 (I+) where the flexible γ-CyD macrocycle folds to embrace the aglycone part. The shape of the fluorescence spectra (Fig. 4B) is consistent with both Ib+ and I+ structures. The quantum yields and the lifetimes in both 1:1 and 2:1 complexes appear to be little modified with respect to those of the free molecule in agreement with the existence of structures with a large exposure of the dihydroxy-anthraquinone part to solvent. MM and MD simulations evidence the daunosamine moiety can also interact favourably with the γ-CyD macrocycle from the secondary rim side. The sugar unit penetrates more deeply into the cavity when the amino group is not protonated (compare structure II and II+ in Fig. 6). Thus the calculations support that two γ-CyD units can be associated to the aglycone and the daunosamine moieties in the 2:1 complex.
The positive–negative split dichroic signal in the 420–580 nm region, due to the presence of the π,π* transitions of the DOX dimer, is maintained in the 1:2 and 2:2 complexes, indicating γ-CyD is not capable of disrupting the stacking interaction of the aromatic chromophores of DOX in either parallel or antiparallel arrangement. The pH effects on the UV-Vis absorption spectrum (Fig. 2) demonstrate γ-CyD strongly perturbs the acid–base equilibria of DOX in the “dimer” concentration regime, both those of the aglycone moiety, more clearly reflected in the spectral changes at λ > 500 nm, and likely also that of the daunosamine. Considering the experimental conditions, where the 2:2 complex largely predominates, and the geometries proposed for the DOX dimer (Scheme 3)30γ-CyD might access both the aglycone pair and the amino sugar tails. The possibility for the latter complexation mode is supported by structures II and II+ in Fig. 6.
We finally remark that CD spectra of the higher order γ-CyD complexes, molecular modeling results and pH effects on electronic absorption consistently support the conclusion that primary binding involves the aglycone part whereas secondary binding involves the daunosamine moiety.
Acknowledgements
We thank the FP7 PEOPLE-ITN project n. 237962-CYCLON for the funding of the research. We also thank Dr Daniele Cesini for his help and continuous support on the grid computing system. We also wish to acknowledge the Distributed Unified Computing for Knowledge (DUCK) cooperation on porting MD applications to the Comput-ER infrastructure (www.comput-er.it).References
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS.
- D. Dal Ben, M. Palumbo, G. Zagotto, G. Capranico and S. Moro, Curr. Pharm. Des., 2007, 13, 2766–2780 CrossRef CAS.
- E. Cukierman and D. R. Khan, Biochem. Pharmacol., 2010, 80, 762–770 CrossRef CAS.
- M. Yokoyama, Expert Opin. Drug Delivery, 2010, 7, 145–158 CrossRef CAS.
- T. Lammers, Adv. Drug Delivery Rev., 2010, 62, 203–230 CrossRef CAS.
- R. Haag, Angew. Chem., Int. Ed., 2004, 43, 278–282 CrossRef CAS.
- M. M. Yallapu, S. P. Foy, T. K. Jain and V. Labhasetwar, Pharm. Res., 2010, 27, 2283–2295 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- T. Betancourt, B. Brown and L. Brannon-Peppas, Nanomedicine, 2007, 2, 219–232 CrossRef CAS.
- T. Nakanishi, S. Fukushima, K. Okamoto, M. Suzuki, Y. Matsumura, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 2001, 74, 295–302 CrossRef CAS.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS.
- Y. Chen and Y. Liu, Chem. Soc. Rev., 2010, 39, 495–505 RSC.
- J. X. Zhang and P. X. Ma, Nano Today, 2010, 5, 337–350 CrossRef CAS.
- F. van de Manakker, T. Vermonden, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2009, 10, 3157–3175 CrossRef CAS.
- E. Bilensoy and A. A. Hincal, Expert Opin. Drug Delivery, 2009, 6, 1161–1173 CrossRef CAS.
- S. Daoud-Mahammed, P. Couvreur, K. Bouchemal, M. Cheron, G. Lebas, C. Amiel and R. Gref, Biomacromolecules, 2009, 10, 547–554 CrossRef CAS.
- S. Daoud-Mahammed, C. Ringard-Lefebvre, N. Razzouq, V. Rosilio, B. Gillet, P. Couvreur, C. Amiel and R. Gref, J. Colloid Interface Sci., 2007, 307, 83–93 CrossRef CAS.
- R. Gref, C. Amiel, K. Molinard, S. Daoud-Mahammed, B. Sebille, B. Gillet, J. C. Beloeil, C. Ringard, V. Rosilio, J. Poupaert and P. Couvreur, J. Controlled Release, 2006, 111, 316–324 CrossRef CAS.
- M. E. Davis, Adv. Drug Delivery Rev., 2009, 61, 1189–1192 CrossRef CAS.
- J. Cheng, K. T. Khin and M. E. Davis, Mol. Pharmaceutics, 2004, 1, 183–193 CrossRef CAS.
- H. Tanaka, K. Kominato, R. Yamamoto, T. Yoshioka, H. Nishida, H. Tone and R. Okamoto, Journal of Antibiotics, 1994, 47, 1025–1029 CAS.
- A. Al-Omar, S. Abdou, L. De Robertis, A. Marsura and C. Finance, Bioorg. Med. Chem. Lett., 1999, 9, 1115–1120 CrossRef CAS.
- P. Y. Grosse, F. Bressolle, P. Vago, J. Simony-Lafontaine, M. Radal and F. Pinguet, Anticancer Res., 1998, 18, 379–384 CAS.
- P. Y. Grosse, F. Bressolle and F. Pinguet, Eur. J. Cancer, 1998, 34, 168–174 CrossRef CAS.
- P. Y. Grosse, F. Bressolle and F. Pinguet, Br. J. Cancer, 1998, 78, 1165–1169 CrossRef CAS.
- P. Y. Grosse, F. Bressolle and F. Pinguet, Cancer Chemother. Pharmacol., 1997, 40, 489–494 CrossRef CAS.
- M. Menozzi, L. Valentini, E. Vannini and F. Arcamone, J. Pharm. Sci., 1984, 73, 766–770 CrossRef CAS.
- M. M. L. Fiallo, H. Tayeb, A. Suarato and A. Garnier-Suillerot, J. Pharm. Sci., 1998, 87, 967–975 CrossRef CAS.
- P. Agrawal, S. K. Barthwal and R. Barthwal, Eur. J. Med. Chem., 2009, 44, 1437–1451 CrossRef CAS.
- N. Husain, T. T. Ndou, A. M. Delapena and I. M. Warner, Appl. Spectrosc., 1992, 46, 652–658 CrossRef CAS.
- O. Bekers, J. J. Kettenesvandenbosch, S. P. Vanhelden, D. Seijkens, J. H. Beijnen, A. Bult and W. J. M. Underberg, J. Inclusion Phenom. Mol. Recognit. Chem., 1991, 11, 185–193 CrossRef CAS.
- O. Bekers, J. H. Beijnen, B. J. Vis, A. Suenaga, M. Otagiri, A. Bult and W. J. M. Underberg, Int. J. Pharm., 1991, 72, 123–130 CrossRef CAS.
- O. Bekers, J. H. Beijnen, M. Otagiri, A. Bult and W. J. M. Underberg, J. Pharm. Biomed. Anal., 1990, 8, 671–674 CrossRef CAS.
- V. Monnaert, D. Betbeder, L. Fenart, H. Bricout, A. M. Lenfant, C. Landry, R. Cecchelli, E. Monflier and S. Tilloy, J. Pharmacol. Exp. Ther., 2004, 311, 1115–1120 CrossRef CAS.
- L. Gallois, M. Fiallo and A. Garnier-Suillerot, Biochim. Biophys. Acta, Biomembr., 1998, 1370, 31–40 CrossRef CAS.
- I. Manet, F. Manoli, B. Zambelli, G. Andreano, A. Masi, L. Cellai and S. Monti, Phys. Chem. Chem. Phys., 2011, 13, 540–551 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, ed.Handbook of Photochemistry, CRC Taylor & Francis, Boca Raton, FL33487–2742, pag. 574 2006 Search PubMed.
- K. Harata, Bull. Chem. Soc. Jpn., 1987, 60, 2763–2767 CrossRef CAS.
- Gaussian 09, Revision A.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- G. Raffaini and F. Ganazzoli, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 683–688 CrossRef CAS.
- AMBER 11, D. A. Case, T. A. Darden, T. E. Cheatham, III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvai, K. F. Wong, F. Paesani, J. Vanicek, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, ( 2010), University of California, San Francisco.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- B. Samori, A. Rossi, I. D. Pellerano, G. Marconi, L. Valentini, B. Gioia and A. Vigevani, J. Chem. Soc., Perkin Trans. 2, 1987, 1419–1426 RSC.
- V. Rizzo, C. Battistini, A. Vigevani, N. Sacchi, G. Razzano, F. Arcamone, A. Garbesi, F. Colonna, M. L. Capobianco and L. Tondelli, J. Mol. Recognit., 1989, 2, 132–141 CrossRef CAS.
- A. Walter, H. Schutz and E. Stutter, Int. J. Biol. Macromol., 1983, 5, 351–355 CrossRef CAS.
- P. Job, Ann. Chim., 1928, 9, 113–203 CAS.
- L. Messori, C. Temperini, F. Piccioli, F. Animati, C. Di Bugno and P. Orioli, Bioorg. Med. Chem., 2001, 9, 1815–1825 CrossRef CAS.
- R. Kiraly and R. B. Martin, Inorg. Chim. Acta, 1982, 67, 13–18 CrossRef CAS.
- K. K. Karukstis, E. H. Z. Thompson, J. A. Whiles and R. J. Rosenfeld, Biophys. Chem., 1998, 73, 249–263 CrossRef CAS.
- Similar lifetimes were also found with DOX 1.6 × 10-4 M in both absence and presence of γ-CyD 1.6 × 10-2 M (DOX > 96% complexed in 2:2 stoichiometry).
- S. Monti and S. Sortino, Chem. Soc. Rev., 2002, 31, 287–300 RSC.
- A. Andreoni, E. J. Land, V. Malatesta, A. J. McLean and T. G. Truscott, Biochim. Biophys. Acta, Gen. Subj., 1989, 990, 190–197 CrossRef CAS.
- The spectra of the complexes are characterized by positive bands in the visible, as expected for monomeric DOX. However in the 1:1 species a slightly negative CD at 550 nm is likely due to some 1:2 complex, not considered in the analysis neglecting the DOX dimer fraction present in solution.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra01221a/ |
| This journal is © The Royal Society of Chemistry 2012 |