Ruthenium dye functionalized gold nanoparticles and their spectral responses†
Linda
Zedler
a,
Frank
Theil
ab,
Andrea
Csáki
b,
Wolfgang
Fritzsche
b,
Sven
Rau
c,
Michael
Schmitt
a,
Jürgen
Popp
ab and
Benjamin
Dietzek
*ab
aInstitute of Physical Chemistry and Abbe Center of Photonics, Jena, Germany
bInstitute for Photonic Technology, Jena, Germany. E-mail: Benjamin.Dietzek@ipht-jena.de
cInstitute of Inorganic Chemistry, Ulm University, Germany
First published on 10th April 2012
Abstract
The functionalization of metal nanoparticles provides access to materials with unique chemical and physical properties for a wide range of applications, e.g. in catalysis, optoelectronics and material science. However, the fundamental light induced charge transfer processes and electronic interactions at the functional nanoparticle–molecule interface, which constitute these unique properties, are not yet fully understood. In this work monodisperse spherical gold nanoparticles functionalized by the photoactive ruthenium dyes (Bu4N)2[Ru(dcbpyH)2(NCS)2], (Bu4N)3[Ru(tcterpy)(NCS)3] and [Ru(dnbpy)(dcbpyH2)(NCS)2] (Bu4N = tetrabutylammonium, dcbpyH = 2,2′-bipyridyl-4,4′-dicarboxylato, NCS = isothiocyanato, tcterpy = 2,2′:6′,6′′-terpyridyl-4,4′,4′′-tricarboxylato, dnbpy = 2,2′-bipyridyl-4,4′-dinonyl) known as N719, N749 and Z907, and [Ru(tbbpy)2(tpphz)](PF6)2 (tbbpy = 4,4′-butyl-2,2′-bipyridine, tpphz = tetrapyridophenazine, PF6 = hexafluorophosphate) (Ru) were synthesized in aqueous solution applying a conjugation and a phase transfer reaction approach, respectively. The functionalized nanoparticles obtained were analyzed by UV-vis spectroscopy, TEM imaging and Raman spectroscopic techniques in order to investigate the molecular structures of the photoactive ruthenium dyes at the gold surface. The results indicate, that the dyes' fully conjugated electronic structure and therefore, their photophysical properties, are preserved or only slightly altered upon binding to the surface of the gold nanoparticles, which potentially allows for rapid and efficient transport of charges to the nanoparticles after photoexcitation.
Introduction
Nanostructured materials have drawn a vast amount of attention in various fields of research during the last decades, due to their unique optical, catalytic and electronic properties.1–3 This development includes nanomaterials composed of metals,4 semiconductors,5 polymers6 but also composite and alloy materials.4 Metal nanoparticles (NPs), and in particular silver and gold nanoparticles (AuNPs), have attracted significant interest, because they combine large structural variability and extraordinary physical properties in comparison to the respective bulk materials.7 These unique properties originate from their small size and large surface-to-volume ratio.8,9 In addition, the physical and chemical properties of NPs can be tuned by surface functionalization10 giving rise to numerous applications, e.g. biological “cloaking” of nanostructured materials for medical research11,12 or novel spectroscopic biosensing approaches at the nanoscale.13 Metal NPs can be functionalized by a wide range of molecules due to the high affinity of atoms such as sulfur,14 nitrogen15 and phosphorus16 to diverse metal surfaces. Thus, the design of functional nanoparticle–molecular interfaces has become a field of intense research.17–22 Semiconductor NPs modified with transition metal dyes have attracted interest as the main compounds of dye-sensitized solar cells.23–25 Classically, such solar cells are based on a ruthenium polypyridyl dye for sensitizing nanostructured TiO2 films, converting visible light into electricity. Several transition metal complexes have been employed as photosensitizers, for example commercially available dyes such as N719, N749 and Z907 (see Fig. 1).26 As a common feature these dyes carry a carboxyl-functionalized polypyridine ligand. The carboxyl groups provide efficient binding to the surface of the NPs and support injection of electrons from the photo-excited ruthenium dye into the conducting band of the TiO2.27 The dyes N719, N749 and Z907 not only exhibit carboxyl groups for binding to semiconductor surfaces, but also possess isothiocyanate ligands with free sulfur atoms that have a high affinity to metals. Although providing beneficial features for applications in photonics, electronics, material science or catalysis7 gold nanoparticle–molecular interfaces with photoactive molecular species—in particular ruthenium polypyridyl dyes—have only been sparsely investigated to date.14,28 In particular, the incorporation of photocatalytic features can increase the impact of these systems even though this represents a rather challenging approach due to the additional directional charge transfer to a catalytic center. Nonetheless, the successful realization of such concepts promises to open a synthetic doorway for the design of, e.g. supramolecular systems for efficient hydrogen generation.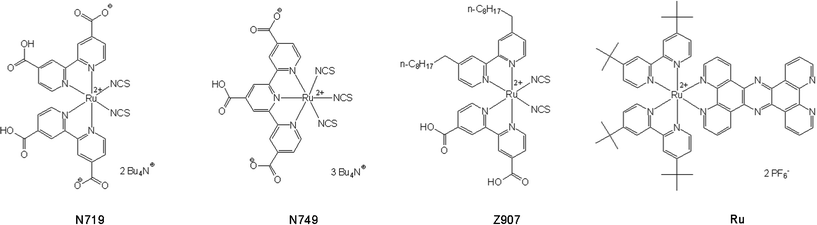 | ||
| Fig. 1 Molecular structure of the ruthenium complexes N749, N719, Z907 and Ru. | ||
This study represents a systematic investigation of the interface between gold NPs and various photoactive ruthenium dyes. N749, N719 and Z907 were attached to the surface of AuNPs to prove that adsorption occurred via the isothiocyanate group and to examine the orientations of the molecules with respect to the metal surface. While N719 functionalized metal NPs have already been synthesized in organic solvents, their stabilization in aqueous solution could not be realized.28 However, an aqueous environment is advantageous for many potential applications, especially in catalysis and obviously for biomedical applications. To the best of our knowledge, the synthesis of N749, N719 and Z907 functionalized AuNPs in aqueous solution is reported here for the first time.
Furthermore, this study demonstrates the immobilization of Ru (see Fig. 1) on the surface of metal NPs. Ru consists of a tetrapyridophenanzine (tpphz) bridging ligand and a ruthenium(II) photocenter and is known to be a light absorbing moiety in an intramolecular photocatalyst reducing protons to H2 under irradiation.29 The molecular photocatalyst, in which the uncoordinated phenantroline sphere of Ru is coordinated to a PdCl2 group has been intensively studied with resonance Raman (RR) and ultrafast spectroscopy30–34 in order to understand the light induced photochemical processes underlying the catalyst’s performance. A promising option to increase the efficiency of such a system might be the use of NPs as a catalytic center instead of a single palladium ion.29 The use of NPs has the potential to overcome two major obstacles of the previously reported approach: in the dinuclear photocatalytic system two electrons need to be generated by photoexcitation of one chromophore and transferred to the catalytic center to produce hydrogen. This is a process of low probability and serious spatial constraints, since photoactivation and reducible hydrogen ions have to meet at the same point in space and time. These constraints are relaxed in nanoparticle systems. First, multiple electrons on the NPs can be photogenerated by different photoactive dye molecules on the NP. Second, the conductive and catalytically active NP surface provides much more area for catalysis and enables the fast distribution of electrons across the NPs. From these viewpoints, we applied a facile method to anchor Ru onto the three dimensional surface of AuNPs, which was ultimately intended to increase the catalytic efficiency.
For spectroscopic investigations of these novel AuNPs the strong surface plasmon resonance absorption band of AuNPs allows optical characterization in the visible region.35 A powerful tool delivering detailed insights into structure related molecular properties is surface-enhanced Raman scattering (SERS).36 We utilized SERS to investigate ruthenium complexes at the surface of the metal NPs. Detection of the molecules' SERS spectra does not only provide proof of binding, e.g. formation of the functional nanoparticle–molecular interface, but also information on the structure of the interface, e.g. the orientation of the adsorbate and its interaction with the nanoparticle's surface.
In summary we report on the synthesis of AuNPs functionalized by photoactive ruthenium dyes with ligands of different natures in an aqueous environment and their analysis by Raman enhancement techniques to investigate the molecular structures of typical photoactive ruthenium dyes in a functional nanoparticle–molecular interface.
Experimental
Synthesis of functionalized AuNPs
The ruthenium complexes (Bu4N)2[Ru(dcbpyH)2(NCS)2], (Bu4N)3[Ru(tcterpy)(NCS)3] and [Ru(dnbpy)(dcbpyH2)(NCS)2] (Bu4N = tetrabutylammonium, dcbpyH = 2,2′-bipyridyl-4,4′-dicarboxylato, NCS = isothiocyanato, tcterpy = 2,2′:6′,6′′-terpyridyl-4,4′,4′′-tricarboxylato, dnbpy = 2,2′-bipyridyl-4,4′-dinonyl) known as N719, N749 and Z907 (see Fig. 1) were purchased from Solaronix SA (Aubonne, Switzerland) and used without further purification. The dyes were immobilized on monodisperse spherical AuNPs of 30 and 60 nm diameter by conjugation. The well studied, yet structurally different ruthenium dye,29,30 [Ru(tbbpy)2(tpphz)](PF6)2 (tbbpy = 4,4′-butyl-2,2′-bipyridine, tpphz = tetrapyridophenazine, PF6 = hexafluorophosphate) (Ru) (see Fig. 1), was bound to colloidal gold by a phase transfer reaction (PTR).There are different reasons for the fact that two synthesis strategies had to be applied. First, N719, N749 and Z907 most likely bind to the surface of AuNPs with the sulfur atoms of the isothiocyanate ligands, which have a high affinity to gold.37Ru cannot employ this binding modality. Nevertheless, it offers the bidentate phenantroline ligand for binding to the gold surface using the lone electron pairs of the nitrogen atoms. Since nitrogen has a much smaller affinity to gold than sulfur the binding is likewise weaker than for the commercially available dyes. Secondly, the main difference of both synthetic methods, as will be presented in the section “UV-Vis and transmission electron microscopic (TEM) imaging” is the distinct mean diameter of the NPs. Binding via the lone electron pairs of the nitrogen atoms of the phenantroline moieties of the tetrapyridophenanzine (tpphz) ligand38 is sterically demanding, thus, and thus favours binding to smaller particles obtained by PTR. Owing to the smaller radius of the NPs the 270 pm wide binding pocket of the tpphz ligand33 firmly binds to the surface of an AuNP. Indeed a 6 nm AuNP is 20 times larger than the tpphz binding pocket; however, the number of nearest neighbours of a single gold atom at the surface is distinctly smaller than for a 30 nm AuNP. Thus, a single gold atom is more easily accessed by the binding pocket of the tpphz ligand. In addition, due to the NPs larger surface-to-volume ratio surface atoms exhibit higher binding energies than atoms of the bulk material. Hence, surface functionalization is also energetically beneficial for smaller particles.39
Conjugation
Spherical AuNPs with mean diameters of 30 and 60 nm were synthesized by citrate reduction.40 The colloidal solutions of the citrate stabilized AuNPs were incubated with 5 vol.% aqueous solutions of the dyes N719, N749 and 5 vol.% ethanolic solution in the case of Z907 with molarities of 10 μM. On average, 1 mL conjugate was obtained by mixing 900 μL NP suspension with 50 μL dye solution filled up with 50 μL distilled water. The conjugate was mixed on a rotating tumbler at room temperature for 2 h. Each batch was cleaned by repeated centrifugation and replacing the residual solution with 1 mL distilled water. The obtained functionalized NPs remained stable for months.Phase transfer reaction
AuNPs stabilized by Ru were prepared by adopting the previously reported two-phase extraction method.41,42 In contrast to the literature reports the reaction time for the phase transfer of AuNPs from toluene to aqueous solution was increased to 15 h.For the AuNP synthesis a 30 μM aqueous solution of HAuCl4·3H2O was added to a 50 μM solution of tetraoctylammonium bromide in toluene (4 mL). The transfer of the metal chloride to the toluene phase could be observed by a color change of the organic phase from transparent to orange within a few seconds. A 0.4 M solution of NaBH4 (0.928 μL), freshly prepared in ice water, was then added to the two-phase mixture and stirred for 1 h at room temperature.
For the phase transfer a dye solution of a ratio [AuIII]/[Ru] = 6 was used. 5 mg (46 μmol) of the chloride salt of Ru dissolved in water (10 mL) were added to the stirred toluene solution. The phase transfer was completed within 15 h, which could be observed by a color change of the aqueous phase from orange to dark brown. The suspensions were purified at least three times by centrifugation. After each centrifugation the residual solvent was removed and replaced by distilled water. The obtained nanoparticles remained stable for months.
Materials and methods
TEM imaging
The size distribution and the agglomeration behaviour of the obtained AuNP suspensions were determined by TEM imaging at 80 kV (DSM 960, Zeiss, Jena, Germany).UV-vis spectroscopy
UV-vis absorption spectra of 100 μM dye solutions and functionalized colloidal gold particles were recorded on a Jasco V670 UV-vis spectrometer using the double beam mode and a 1 mm quartz cuvette.Raman spectroscopy
Raman, resonance Raman (RR), and SERS spectra were acquired using various experimental setups. Nonresonant FT-Raman spectra (λexc = 1064 nm) were recorded using a “MultiRAM” (Bruker) with a fibre-coupled diode pumped solid-state laser (DENICAFC LC-3/40, KLASTECH-Karpushko Laser Technologies). The laser power was set to 100 mW. For each spectrum 100 scans were averaged. A single IR 352 objective with a working distance of 16 mm focused the laser on the sample and collected the scattered light. The signal was detected by a nitrogen-cooled Ge-Diode (Bruker D418-T). The spectral resolution was 2 cm−1.The RR and SERS spectra were excited with an Ar ion laser (Coherent) operated at 514 and 458 nm or a frequency doubled Nd-YAG-laser at 532 nm. The laser was focused into a rotating-cell cuvette for RR43 or a 1 cm fluorescence cuvette for SERS measurements. Raman signals were collected at a 90° scattering angle using either a Canon 1.4 50-mm objective or a lens (f = 100 mm) and focused onto the entrance slit of an Acton SpectraPro 2758i spectrometer (entrance slit width of 100 μm, focal length 750 mm, grating 600 mm−1). The Raman signals were detected by a liquid-nitrogen cooled CCD (Princeton Instruments). Sample integrity was ensured by recording absorption spectra before and after the RR experiments.
Results and discussion
In this study AuNPs stabilized by ruthenium dyes in an aqueous environment were synthesized and characterized by Raman spectroscopic techniques to derive structure-related properties of the nanoparticle–molecular interfaces. The ruthenium complexes used for functionalization exhibit unique optical properties, such as a large absorption cross-section throughout the visible spectral range in the cases of N719, N749 and Z907 and favourable intramolecular charge-transfer properties in the case of Ru.29,30 The spectroscopic analysis presented in the following sections aims at investigating whether these molecular optical properties are preserved upon binding onto AuNPs or not. To this end the interaction of the dyes with the metal surface and the novel properties of this functional nanoparticle–molecular interface were examined by UV-vis absorption, TEM imaging, SERS, RR and nonresonant Raman spectroscopy.UV-vis and TEM imaging
The size distribution of the NPs obtained by conjugation or PTR were analyzed by UV-vis spectroscopy and TEM imaging. The maximum of the surface plasmon resonance (SPR) peak in the UV-vis absorption spectra depends on the mean particle diameter:7,39 the absorption maximum shifts to lower wavelengths and the spectral width narrows for decreasing particle diameter. Since this spectral feature also depends on other parameters (e.g. the refractive index of the solvent, particle shape and temperature), the determination of the particles’ mean diameter using UV-vis spectroscopy is not very precise. In addition, functionalization alters the absorption properties of AuNPs due to e.g. the interaction between the ligand field and the surface electron cloud.13 The UV-vis spectrum in Fig. 2A displays the SPR band as expected from the literature7 for AuNPs of 60 nm diameter with a maximum at about 535 nm, stabilized either by citrate or by N749. A redshift of 7 nm (240 cm−1) of the SPR band has been observed upon ligand exchange, indicating the interface modification of the AuNPs (Fig. 2A). In case of Ru anchored on AuNPs a significant redshift by 24 nm (850 cm−1) was observed for the SPR band after the phase transfer (Fig. 2B). This behaviour can be explained by two effects: first, binding of Ru onto the surface of AuNPs causes a different surrounding environment and influences the surface electron cloud; second, the refractive index of the solvent (nwater = 1.33, ntoluene = 1.496) has been shown to induce a shift of the SPR band, according to the Mie theory44 upon transfer of AuNPs from toluene to water.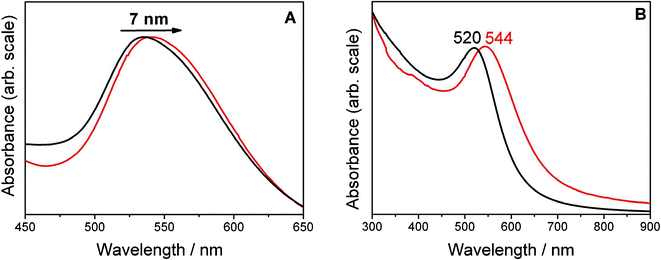 | ||
| Fig. 2 A: Normalized UV-Vis absorption spectra of 60 nm gold colloid stabilized by citrate (black) and N749 (red), the SPR band of the gold colloid shifts markedly by 7 nm upon ligand exchange. B: SPR band of the organosol (black), redshift of the SPR band by 24 nm after PTR (red). | ||
The distinct size distribution was analyzed by TEM imaging (Fig. 3). In the case of conjugation colloidal solutions of about 30 and 60 nm mean diameter with a size distribution of 3 nm (standard deviation) were synthesized. In contrast, the PTR provided much smaller particles (average particle size is 6 ± 1.4 nm) while showing a similar uniform size distribution. The histograms of the size distributions as derived from several TEM images and two sample TEM images are shown in Fig. 3.
 | ||
| Fig. 3 In the upper panels example TEM images and in the lower panels the respective histograms of the NPs obtained by conjugation (A) and phase transfer catalysis (B) are displayed. (A) Shows NPs of 30 nm mean diameter functionalized with Z907. In (B) colloids functionalized with Ru are shown. The mean diameter for the colloid in (B) is 6 ± 1.4 nm and thus much smaller than in the case of A, 28 ± 3 nm. The histograms of the particle diameter of the ruthenium dye coated NPs as derived from multiple TEM images are shown below. | ||
Raman spectroscopic characterization
SERS and RR spectroscopy have been utilized in order to study the binding modality of the ruthenium dyes onto the AuNPs, to analyze the structure and orientation of these complexes on the surface and to identify potential differences compared to the molecular structures in solution. To enable a reliable band assignment of the resonance and surface-enhanced Raman modes of the dye molecules under investigation, nonresonant FT-Raman spectra were recorded for comparison. Furthermore, the nonresonant FT-Raman measurements have been exploited to estimate the magnitude of Raman signal enhancement in both RR and SERS measurements and to characterize the change from the ground state electronic configuration upon excitation.Raman analysis of N719, N749 and Z907
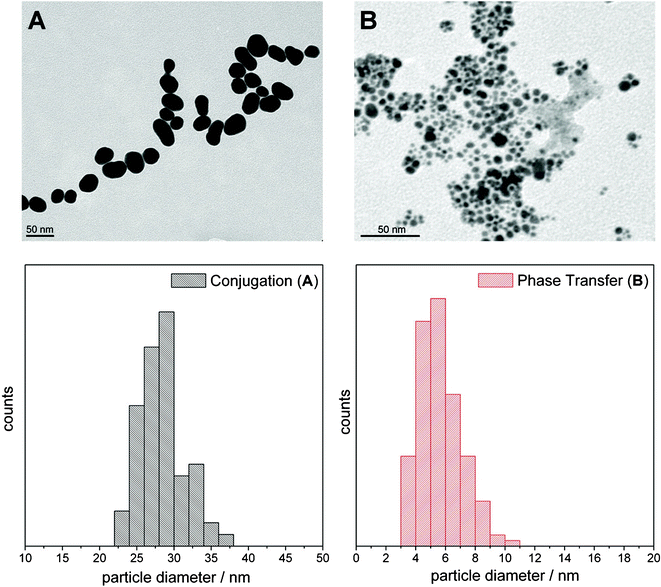
The excitation wavelengths for RR spectroscopy measurements have been chosen to be in resonance with the metal-to-ligand charge transfer (MLCT) absorption band of the dyes in solution (see Fig. 4A). SERS spectra were recorded with an excitation wavelength lying both within the surface plasmon resonance and the MLCT absorption band. | ||
| Fig. 4 UV-vis absorption spectra of 10−4 M solutions of N749, N719, Z907 (A) and Ru (B). The RR excitation laser lines at 532, 514, and 458 nm and the SERS excitation wavelength at 532 nm, in resonance with the dye but also with the surface plasmon resonance of AuNPs, are displayed as vertical lines. | ||
Fig. 5 exemplarily displays the RR, SERS and nonresonant FT-Raman spectra for N749. Both RR and SERS spectra are dominated by modes which can be assigned to the terpyridine (terpy) ligand (terpy—vibrations at 1468, 1521, 1604 cm−1 and less pronounced the ring breathing mode at 1000 cm−1).45Fig. 5A–C show RR spectra recorded for three different concentrations. Based on the S/N ratio of these RR spectra a detection limit of about 10−6 M can be estimated. Furthermore, Fig. 5 shows that the nonresonant FT-Raman spectrum of the pure dye (E) and the RR spectrum of the 10−4 M solution (A) have a comparable S/N ratio, which shows that the nonresonant Raman signal is enhanced by a factor of roughly 104 through resonant MLCT excitation.
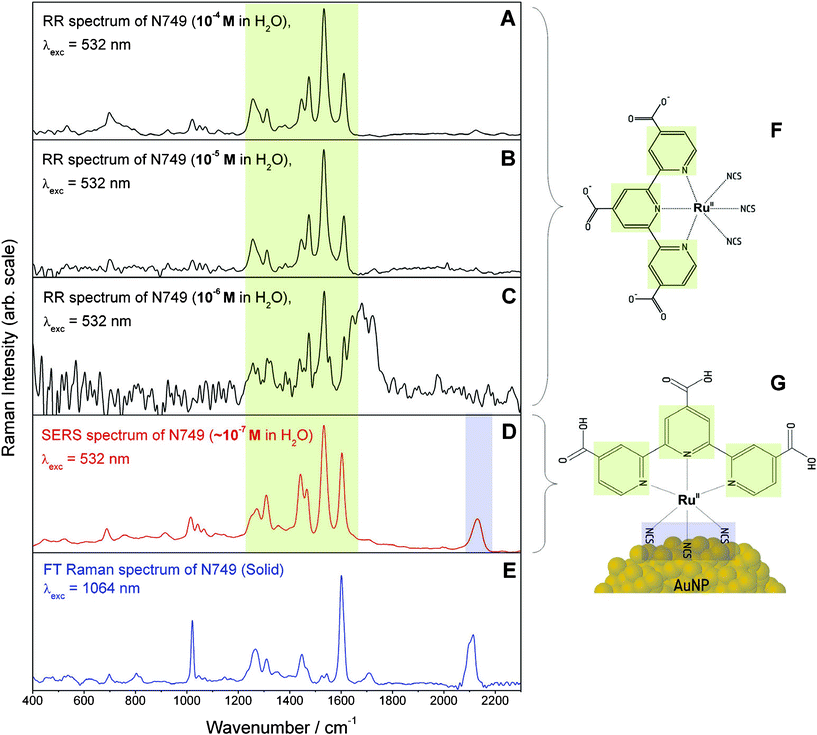 | ||
| Fig. 5 Comparison of RR spectra at 10−4, 10−5 and 10−6 M (A–C), SERS (D) and nonresonant FT-Raman (E) spectra of N749. The broad spectral feature around 1700 cm−1 in panel C is an artefact resulting from subtracting the solvent spectrum. The charge localization on the terpy ligand upon MLCT absorption within the fingerprint spectral region from 1000–1800 cm−1 is marked in green while the binding site via the isothiocyanate group at 2100 cm−1 is highlighted in blue. In panels F and G the structure of the ruthenium dye in solution and the hypothesized binding geometry on the NP surface are depicted. The structural elements are colour coded corresponding to the assignment of the Raman spectral features. | ||
The concentration of the dye on the AuNPs has been estimated to be approximately 10−7 M by assuming a dye monolayer on the surface of 30 nm AuNPs and a concentration of 1010 particles per mL (the area occupied by a dye molecule has been estimated to be about 2 × 10−18 m2). The SERS spectrum shown in Fig. 5D exhibits a similar S/N ratio as the RR spectrum of the 10−4 M dye solution; therefore we conclude that binding the dye to the surface of the AuNPs, i.e. the formation of a functional nanoparticle–molecular interface, leads to a further enhancement by a factor of 103 as compared to RR enhancement.
In order to understand the implications derived from the observed SERS spectrum we will briefly summarize which factors affect the additional SERS enhancement.1,2 Firstly, the SERS effect consists of two contributions, a pure electromagnetic enhancement responsible for most of the total enhancement and an additional chemical enhancement due to the formation of surface complexes with different electronic structure allowing for additional resonance enhancement. The latter one is visible only if the molecule is directly bound to the surface. If the electronic structure of the surface complex is strongly altered, the Raman spectra of such surface complexes also differ significantly from the RR spectra in solution. The longer ranged pure electromagnetic contribution decays more slowly but nevertheless extremely rapidly with distance from the surface. Thus vibrations of molecular bonds in close proximity to the surface are significantly more enhanced than those of structures lying further apart, since the enhancement scales inversely to the 10th power of the distance to the surface for a molecular monolayer. Furthermore the enhancement is also dependent on the orientation with respect to the surface normal. The highest enhancement is achieved for vibrations perpendicular to the surface. According to these properties of SERS enhancement a clear proof of binding to the metal is the observation of the CN-stretching vibration at about 2100 cm−1 due to the isothiocyanate group in the SERS spectrum (Fig. 5D), which—in addition—is the only notable difference between the SERS and RR spectra. Further vibrational bands of this functional group e.g. the Au–S stretching vibration at around 450 cm−1 and the antisymmetric NCS stretching vibration were not observable to support our interpretation for several reasons. The strong reflections from the colloid the background are very inhibiting within the low wavenumber region, hence the Au–S stretching vibration cannot be observed. Second these vibrations are only enhanced by the electromagnetic SERS effect, since there is no evidence for a marked resonance enhancement of these bands. Otherwise they would be observable in the RR spectrum, too. Thus fortunately the symmetric CN vibration is within a silent spectral region enabling clear detection despite relatively weak enhancement. In contrast to previously published SERS investigations of the dyes using AuNP suspensions in organic solvents,28 no bands of the COO-groups are visible in the SERS spectrum (see Fig. 5D). Since surface enhancement drastically depends on the distance between a molecular fragment and the SERS active metal surface, we conclude that N749 is anchored on the AuNPs via the sulfur atoms of the isothiocyanate ligands, which have a high affinity to gold.
In order to characterize the Franck–Condon region of N749, RR and nonresonant FT-Raman spectra were compared to identify which modes are resonantly enhanced i.e. are coupled to the MLCT transition. Such a comparison between Fig. 5A and E clearly shows a selective enhancement of characteristic bands of the terpy ligand at 1468, 1521 and 1604 cm−1 demonstrating that the electronic excitation is localized on this ligand45 (see Fig. 5).
Of particular interest is the investigation of the dye adsorbed on the surface of AuNPs and how its structure differs from the structure of the dye in solution. By comparing the wavenumber positions and relative intensities of the terpy bands in the RR and SERS spectra (Fig. 5A and D), it is obvious that both spectra are very similar. This similarity points towards similar molecular and electronic structures of the unbound N749 and the N749 attached to the AuNP’s surface. Thus, the fully conjugated chromophoric system within N749 is preserved upon binding to the AuNPs, which potentially allows for rapid and efficient transport of charges to the NPs after photoexcitation.
Analogous spectroscopic results have been obtained for colloids stabilized by Z907 and N719. The corresponding spectra are presented in the supplementary information.†
In the following, similar experiments on Ru are discussed in order to investigate, how its electronic properties are affected by immobilization on AuNPs.
Raman analysis of Ru
Fig. 6 depicts the RR, SERS and nonresonant FT-Raman spectra of Ru. It should be noted that in the case of Ru it was not possible to excite RR and SERS spectra with similar excitation wavelengths as used for N719, N749 and Z907. Instead, SERS spectra were recorded with an excitation wavelength of 532 nm assuring resonant excitation conditions within the SPR band of the AuNPs. However, it was not possible to obtain RR spectra for free Ru in solution with 532 nm excitation due to the presence of a strong fluorescence background obscuring the RR information. Thus, RR spectra were recorded in the maximum of the MLCT band at 458 nm (see Fig. 4B) where the RR bands are spectrally well separated from the fluorescence emission. The RR spectrum is characterized by prominent bands of the tpphz ligand at 1601, 1574, 1506, 1450 and 1190 cm−1 and the tbbpy ligand at 1538, 1481, 1317, 1131 and 1028 cm−1.31 This band assignment is based on a comparison between RR and nonresonant FT-Raman spectra (Fig. 6A and C) and is in agreement with previous RR spectroscopic studies and quantum-chemical calculations.32 Similar to N749, the detection limit of Ru with RR is 10−6 M (see Fig. S3†). The SERS spectrum recorded at 532 nm is dominated by bands of both, tbbpy and tpphz ligands, and exhibits a similar S/N ratio as the RR spectrum (Fig. 6B). The concentration of the dye on the AuNPs is estimated to be 10−7 M using the approximations laid out above for N749.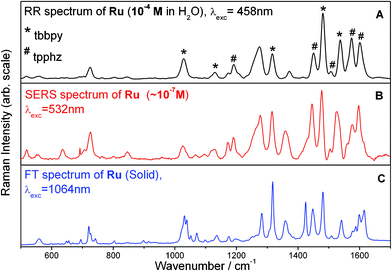 | ||
| Fig. 6 Comparison of RR (A), SERS (B) (Ru stabilized AuNPs obtained from PTR), and FT-Raman (C) spectra for Ru. Raman bands which can be assigned to the tbbpy ligand are marked by a * while tpphz bands are labelled with a #, according to the literature.31 | ||
To verify the formation of a nanoparticle–molecular interface, additional SERS enhancement, i.e. the Raman signal enhancement due to the proximity of the metal surface and not due to the electronic resonance of the excitation wavelength, has to be estimated. Since the S/N ratio of the RR spectra (Fig. 6A) at a concentration of 10−4 M and the SERS spectrum (Fig. 6B) at 10−7 M are similar, the additional SERS enhancement is estimated to be 103. The increased SERS enhancement of modes assigned to the bidentate tpphz ligand as compared to the RR spectrum is a strong indicator for an attachment of Ru to the gold surface via the tpphz ligand. In particular the tpphz bands at 1603 and 1453 cm−1 (Fig. S4, ESI†) exhibit a significantly larger relative intensity in the SERS spectrum than in the RR spectrum. The chelating bidentate tpphz group ensures a strong interaction with the metal surface.
As for N719, N749 and Z907 the determination of structural differences of Ru adsorbed on the surface of AuNPs in comparison to its structure in solution is of importance, since its photocatalytic properties, when coordinated to a catalytic active unit, are strongly dependent on the preservation of the fully conjugated electronic structure of the tpphz ligand upon binding. As pointed out above, the relative intensities of the RR and SERS bands are not identical (see Fig. 6A and B). This discrepancy might be partially explained by the different excitation wavelengths. Since the excitation wavelengths of SERS and RR spectra differ by more than 70 nm, the spectral changes in the band pattern may be due to different resonance enhancement. Such effects were observed for structurally similar complexes.31,33,46 Overall, we do not have sufficient evidence for assuming similar molecular and especially electronic structures of Ru attached to AuNPs and Ru in solution. In order to clarify if the conjugated electronic structure required for rapid and efficient transport of charges after photoexcitation is preserved upon immobilization on AuNPs, RR spectra need to be recorded at an excitation wavelength of 532 nm. However, as mentioned above these experiments failed due to strong fluorescence masking the RR bands of Ru in solution.
Conclusion
Nanostructured materials are promising candidates for a broad range of applications due to their unique optical, catalytic and electronic properties arising from their size and the additional functionalization potential of the large surface area. By modifying the surface of AuNPs exclusive properties of a molecular monolayer can be introduced and combined with the properties and functionalities of the particles. In this contribution, the immobilization of the ruthenium based dyes (Bu4N)2[Ru(dcbpyH)2(NCS)2] (N719), (Bu4N)3[Ru(tcterpy)(NCS)3] (N749), [Ru(dnbpy)(dcbpyH2)(NCS)2] (Z907) and [Ru(tbbpy)2(tpphz)](PF6)2 (Ru) on AuNPs has been demonstrated for the first time in aqueous solution. Due to the different affinity of the various ligands to the gold surface two synthesis strategies have been applied. Phase transfer reaction and conjugation result in very uniform, homogeneous and long term stable AuNPs. In order to analyze the special optical and chemical properties arising from the functional nanoparticle–molecular interface, Raman spectroscopic methods have been employed. N749, N719 and Z907 are bound to AuNPs via the isothiocyanate ligand as concluded from the surface-enhanced Raman scattering (SERS) study. The dyes' fully conjugated electronic structure and therefore also their photochemical properties are preserved, since almost no spectral changes were detected when comparing resonance Raman (RR) of the dyes to SERS spectra of the dyes adsorbed on the surface of AuNPs.In the case of Ru, a well-studied photoactive complex, the comparison between RR and SERS spectra provides evidence that the electronic structure of this ruthenium complex might be slightly changed upon anchoring to AuNPs. However, this issue needs to be further clarified. This and an analysis of a possible catalytic activity of Ru functionalized gold nanoparticles will be the subject of future studies.
The synthesis of ruthenium dye modified gold nanoparticles in watery environment opens the door to designing nanoparticles of unique photochemical properties. Both, the gold nanoparticles and the stabilizing agent can be independently investigated and modified at a molecular level in order to improve potential charge transfer reactions. If the applied synthesis concepts can be transferred to different transition metal colloids of known reactivity or catalytic activity, numerous new applications in the fields of catalysis, electronics, nanotechnology and material science can be addressed.
Acknowledgements
This work was supported by the Studienstiftung des deutschen Volkes, the Fonds der Chemischen Industrie and the Thüringer Ministerium für Bildung, Wissenschaft und Kultur (Grant No. B 514-09049, PhotoMIC). The authors are thankful to Franka Jahn for the TEM measurements.References
- D. G. Schmid, Nanoparticles: from theory to application, Wiley-VCH, 2004 Search PubMed.
- A.-I. Henry, J. M. Bingham, E. Ringe, L. D. Marks, G. C. Schatz and R. P. Van Duyne, Correlated Structure and Optical Property Studies of Plasmonic Nanoparticles, J. Phys. Chem. C, 2011, 115(19), 9291–9305 CAS.
- H. M. Chen and R.-S. Liu, Architecture of Metallic Nanostructures: Synthesis Strategy and Specific Applications, J. Phys. Chem. C, 2011, 115(9), 3513–3527 CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Shape-Controlled Synthesis of Metal Nanocrystals: Simple Chemistry Meets Complex Physics?, Angew. Chem., Int. Ed., 2008, 48(1), 60–103 Search PubMed.
- G. Cao and C. J. Brinker, Annual review of nano research, World Scientific, 2008 Search PubMed.
- S. Thomas, Recent Advances in Polymer Nanocomposites: Synthesis and Characterization, Brill, 2010 Search PubMed.
- M.-C. Daniel and D. Astruc, Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology, Chem. Rev., 2004, 104(1), 293–346 CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Colloidal silver nanoplates. State of the art and future challenges, J. Mater. Chem., 2008, 18, 1724 CAS.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Shape control in gold nanoparticle synthesis, Chem. Soc. Rev., 2008, 37, 1783 CAS.
- H. Oikawa, T. Onodera, A. Masuhara, H. Kasai and H. Nakanishi, New Class Materials of Organic-Inorganic Hybridized Nanocrystals/Nanoparticles, and Their Assembled Micro- and Nano-Structure Toward Photonics, inPolymer Materials, vol. 231, K.-S. Lee and S. Kobayashi, ed., Berlin, Heidelberg: SpringerBerlin Heidelberg, 2009, pp. 147–190 Search PubMed.
- C. Minelli, S. B. Lowe and M. M. Stevens, Engineering Nanocomposite Materials for Cancer Therapy, Small, 2010, 6(21), 2336–2357 CAS.
- D. R. Cooper and J. L. Nadeau, Nanotechnology for in vitro neuroscience, Nanoscale, 2009, 1, 183 CAS.
- A. Csaki, T. Schneider, J. Wirth, N. Jahr, A. Steinbrück, O. Stranik, F. Garwe, R. Müller and W. Fritzsche, Molecular plasmonics: light meets molecules at the nanoscale, Philos. Trans. R. Soc. London, Ser. A, 2011, 369(1950), 3483–3496 CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, Synthesis of thiol-derivatised gold nanoparticles in a two-phase Liquid–Liquid system, J. Chem. Soc., Chem. Commun., 1994,(7), 801 CAS.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Synthesis of Nanosized Gold-Silica Core-Shell Particles, Langmuir, 1996, 12(18), 4329–4335 Search PubMed.
- A. Moores, F. Goettmann, C. Sanchez and P. Le Floch, Phosphinine stabilised gold nanoparticles; synthesis and immobilisation on mesoporous materials, Chem. Commun., 2004, 2842 CAS.
- J. C. Rubim, Surface-Enhanced Raman Spectroscopic (SERS and FT-SERS) Investigation of the Complex Ion [Fe2(CN)10L]6− (L = 4,4′-Bipyridine and Pyrazine) Adsorbed on Silver and Gold Electrodes, J. Phys. Chem., 1995, 99(1), 345–355 CAS.
- T. Huang and R. W. Murray, Quenching of [Ru(bpy)3]2+ Fluorescence by Binding to Au Nanoparticles, Langmuir, 2002, 18(18), 7077–7081 CAS.
- P. Corio, G. F. S. Andrade, I. C. N. Diógenes, I. S. Moreira, F. C. Nart and M. L. A. Temperini, Characterization of the [Ru(CN)5(pyS)]4− ion complex adsorbed on gold, silver and copper substrates by surface-enhanced Raman spectroscopy, J. Electroanal. Chem., 2002, 520(1–2), 40–46 CAS.
- C. R. Mayer, E. Dumas and F. Sécheresse, Size controlled formation of silver nanoparticles by direct bonding of ruthenium complexes bearing a terminal mono- or bi-pyridyl group, Chem. Commun., 2005,(3), 345 CAS.
- A. J. Hallett, P. Christian, J. E. Jones and S. J. A. Pope, Luminescent, water-soluble gold nanoparticles functionalised with 3MLCT emitting rhenium complexes, Chem. Commun., 2009,(28), 4278 CAS.
- A. Kotiaho, Photoinduced Charge and Energy Transfer in Phthalocyanine-Functionalized Gold Nanoparticles, J. Phys. Chem. C, 2010, 114(1), 162–168 CAS.
- B. O'Regan and M. Grätzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films, Nature, 1991, 353(6346), 737–740 CAS.
- L. M. Peter, The Grätzel Cell: Where Next?, J. Phys. Chem. Lett., 2011, 2(15), 1861–1867 CAS.
- A. Listorti, B. O'Regan and J. R. Durrant, Electron Transfer Dynamics in Dye-Sensitized Solar Cells, Chem. Mater., 2011, 23(15), 3381–3399 CAS.
- M. Grätzel, Solar Energy Conversion by Dye-Sensitized Photovoltaic Cells, Inorg. Chem., 2005, 44(20), 6841–6851 Search PubMed.
- G. Benkö, J. Kallioinen, J. E. I. Korppi-Tommola, A. P. Yartsev and V. Sundström, Photoinduced Ultrafast Dye-to-Semiconductor Electron Injection from Nonthermalized and Thermalized Donor States, J. Am. Chem. Soc., 2002, 124(3), 489–493 Search PubMed.
- C. Pérez León, L. Kador, B. Peng and M. Thelakkat, Influence of the Solvent on the Surface-Enhanced Raman Spectra of Ruthenium(II) Bipyridyl Complexes, J. Phys. Chem. B, 2005, 109(12), 5783–5789 Search PubMed.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, A Supramolecular Photocatalyst for the Production of Hydrogen and the Selective Hydrogenation of Tolane, Angew. Chem., Int. Ed., 2006, 45(37), 6215–6218 CAS.
- S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. González, M. Schmitt, S. Rau and J. Popp, Photochemical Fate: The First Step Determines Efficiency of H2 Formation with a Supramolecular Photocatalyst, Angew. Chem., Int. Ed., 2010, 49(23), 3981–3984 CAS.
- S. Tschierlei, B. Dietzek, M. Karnahl, S. Rau, F. M. MacDonnell, M. Schmitt and J. Popp, Resonance Raman studies of photochemical molecular devices for multielectron storage, J. Raman Spectrosc., 2008, 39(5), 557–559 CAS.
- S. Tschierlei, M. Karnahl, C. Kuhnt, F. W. Heinemann, M. Schmitt, S. Rau, J. Popp and B. Dietzek, Photophysics of an Intramolecular Hydrogen Evolving Ru-tpphz-Pd Photocatalyst, Chem.–Eur. J., 2009, 15(31), 7678–7688 CAS.
- M. Karnahl, S. Tschierlei, C. Kuhnt, B. Dietzek, M. Schmitt, J. Popp, M. Schwalbe, S. Krieck, H. Görls, F. W. Heinemann and S. Rau, Synthesis and characterization of regioselective substituted tetrapyridophenazine ligands and their Ru(II) complexes, Dalton Trans., 2010, 39(9), 2359 CAS.
- M. Karnahl, C. Kuhnt, F. Ma, A. Yartsev, M. Schmitt, B. Dietzek, S. Rau and J. Popp, Tuning of Photocatalytic Hydrogen Production and Photoinduced Intramolecular Electron Transfer Rates by Regioselective Bridging Ligand Substitution, ChemPhysChem, 2011, 12(11), 2101–2109 CAS.
- U. Kreibig, Optical properties of metal clusters, Berlin, New York: Springer, 1995 Search PubMed.
- D. Cialla, A. März, R. Böhme, F. Theil, K. Weber, M. Schmitt and J. Popp, Surface enhanced Raman spectroscopy (SERS): progress and trends, Anal. and Bioanal. Chem., 2012, 403(1), 27–54 CAS.
- L. H. Dubois and R. G. Nuzzo, Synthesis, Structure, and Properties of Model Organic Surfaces, Annu. Rev. Phys. Chem., 1992, 43(1), 437–463 CAS.
- Y. Peng, Z. Niu, W. Huang, S. Chen and Z. Li, Surface-Enhanced Raman Scattering Studies of 1,10-Phenanthroline Adsorption and Its Surface Complexes on a Gold Electrode, J. Phys. Chem. B, 2005, 109(21), 10880–10885 CAS.
- Y. Sun, S. K. Gray and S. Peng, Surface chemistry: a non-negligible parameter in determining optical properties of small colloidal metal nanoparticles, Phys. Chem. Chem. Phys., 2011, 13(25), 11814–11826 CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, A study of the nucleation and growth processes in the synthesis of colloidal gold, Discuss. Faraday Soc., 1951, 11(0), 1951 Search PubMed.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, Synthesis and reactions of functionalised gold nanoparticles, J. Chem. Soc., Chem. Commun., 1995,(16), 1655–1656 CAS.
- C. R. Mayer, E. Dumas and F. Sécheresse, 1,10-Phenanthroline and 1,10-phenanthroline-terminated ruthenium(II) complex as efficient capping agents to stabilize gold nanoparticles: Application for reversible aqueousâ organic phase transfer processes, J. Colloid Interface Sci., 2008, 328(2), 452–457 CAS.
- W. Kiefer and H. J. Bernstein, A Cell for Resonance Raman Excitation with Lasers in Liquids, Appl. Spectrosc., 1971, 25(4), 500–501 CAS.
- S. Underwood and P. Mulvaney, Effect of the Solution Refractive Index on the Color of Gold Colloids, Langmuir, 1994, 10(10), 3427–3430 CAS.
- G. C. Vougioukalakis, Terpyridine- and 2,6-dipyrazinylpyridine-coordinated ruthenium(II) complexes: Synthesis, characterization and application in TiO2-based dye-sensitized solar cells, J. Photochem. Photobiol., A, 2010, 214(1), 22–32 CAS.
- M. Schwalbe, M. Karnahl, S. Tschierlei, U. Uhlemann, M. Schmitt, B. Dietzek, J. Popp, R. Groake, J. G. Vos and S. Rau, The switch that wouldn't switch—unexpected luminescence from a ruthenium(II)-dppz-complex in water, Dalton Trans., 2010, 39(11), 2768–2771 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01248k |
| This journal is © The Royal Society of Chemistry 2012 |