Room-temperature columnar mesophases of nickel-bis(dithiolene) metallomesogens†
Sisir
Debnath
a,
Hassan F.
Srour
a,
Bertrand
Donnio
*b,
Marc
Fourmigué
a and
Franck
Camerel
*a
aSciences Chimiques de Rennes, Université de Rennes 1, UMR CNRS 6226, Campus de Beaulieu, 35042, Rennes, France. E-mail: franck.camerel@univ-rennes1.fr
bInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), Université De Strasbourg-CNRS (UMR 7504), 23 rue du Loess, BP 43, 67034, Strasbourg Cedex 2, France. E-mail: bdonnio@ipcms.u-strasbg.fr
First published on 10th April 2012
Abstract
Discotic nickel dithiolene complexes are prepared by sulfuration of original benzil proligands appended with gallate fragments carrying long carbon chains (n = 8, 12, 16), separated from the dithiolene core by ester or amide linkers. The electronic properties of these functional nickel dithiolene complexes were studied by cyclic voltammetry and UV-vis spectroscopy. The thermotropic properties were investigated by a combination of POM observations, DSC analysis and X-ray diffraction experiments. The neutral dithiolene complexes, denoted CnesterNi and CnamideNi, all exhibit columnar phases over large temperature ranges including room temperature. CnesterNi complexes form columnar mesophases of rectangular (pseudo-hexagonal) and hexagonal symmetry whereas the amide linkage in CnamideNi complexes strongly stabilizes hexagonal columnar mesophases far below room temperature, as illustrated by C12amideNi which displays a Colh mesophase from −18 up to +177 °C. The role of hydrogen bonding between the amide functions in the stabilisation of the mesophases in CnamideNi complexes and their benzil precursors was confirmed by IR spectroscopy. Moreover, the C8amideNi compound exhibits a cubic phase, stable over ∼50 °C, from 155 to 203 °C. Models for the arrangements within the different columnar mesophases are proposed and discussed.
Introduction
Supramolecular chemistry,1 in which non-covalent interactions drive the formation of elaborate and exotic superstructures, is an elegant way to organize small molecules into highly functional architectures of potential interest in life and material sciences.2 Among the diversity of materials able to self-assemble into organized superstructures at the micro- and macroscopic scale, liquid-crystalline materials (LCs) continuously emerge as attractive candidates to form active layers, especially in the fields of optics and electronics.3 The family of discotic liquid crystals is a particularly attractive class of materials since they possess the ability to self-organize into various ordered columnar structures. The intriguing large and anisotropic charge mobilities arising from the self-assembly of aromatic molecules into stacked structures make these systems attractive for their applications in photonic and optoelectronic devices,4 such as field-effect transistors (FETs),5 organic light emitting diodes (OLEDs),6 organic light emitting transistors (OLETs)7 and solar cells.8 Additional advantages of columnar discotic liquid crystals are their ease of processing (via solution techniques), the possibility of alignment by shear forces or by application of electrical or magnetic fields, and their capacity for self-healing.Besides, metallomesogens are also fascinating materials combining the fluidity and the self-organisation properties of liquid crystals with the unique magnetic and electronic properties of metal ions, which expand the range of technological applications. Such materials thus permit to organize and orient the specific properties of metal complexes into matrix displaying fast orientational response to external stimuli at the nanoscale. In addition, metal ions offer multiple coordination modes, which can allow the emergence of novel molecular architectures or mesophases. In this respect, the design and synthesis of discotic metallomesogens prepared from protomesogenic ligands having oxygen or nitrogen atoms for metal complexation are legion and have been widely explored.9 Strangely however, metal-complexation of sulfur-based ligands still remains scarce and only a few liquid crystalline disc-like materials have been obtained with functional dithiooxamides,10 dithiocarboxylates11 and dithiolene derivatives.12
Metal-bis(dithiolene) and their derivatives are strong near-IR absorbers with unique electrochemical properties. In their neutral state, they display high absorption coefficients (around 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) in a wide range of NIR absorption maxima that are tunable from 900 to 1600 nm by the judicious combination of metal centre and dithiolene substituents.13 The only known liquid crystalline columnar mesophases were reported by Otha for octasubstituted dithiolene nickel complexes (n = 1–12), monotropic for the short chain-length homologues (n = 2–4), and enantiotropic for longer ones (n > 5) (molecules A).12
000 M−1 cm−1) in a wide range of NIR absorption maxima that are tunable from 900 to 1600 nm by the judicious combination of metal centre and dithiolene substituents.13 The only known liquid crystalline columnar mesophases were reported by Otha for octasubstituted dithiolene nickel complexes (n = 1–12), monotropic for the short chain-length homologues (n = 2–4), and enantiotropic for longer ones (n > 5) (molecules A).12
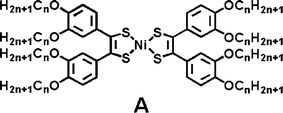
The identity of the mesophase was unequivocally determined by X-ray diffraction to be Colh while high electron mobilities were reported for these mesomorphic Ni-bis(dithiolene) complexes with π-stacked columnar structures.14 Recently, we also described the analogous but paramagnetic octasubstituted gold dithiolene complex and demonstrated that the extent of intermolecular magnetic interactions could be finely controlled at the crystal-to-mesophase transition to generate marked magnetic hysteresis.15 Since the pioneered work of Ohta, a few attempts to develop new dithiolene ligands able to generate mesophases after metal-complexation have been made, but remained unsuccessful.16 In addition, the only known columnar mesophases observed with the octasubstituted dithiolene complexes12,15 are stable between 70 and 110 °C which render these liquid crystalline materials unsuitable for practical optoelectronic applications at room temperature.
Room-temperature liquid crystals are materials with melting points below 25 °C. To decrease the melting point of a mesogenic material, one strategy is to increase the volume fraction of the long flexible carbon chains around the rigid core.9b,17 We have tried to introduce 12 carbon chains around the nickel-bis(dithiolene) core (B) by the synthetic route described by Ohta11,12 for molecules A but the syntheses of the benzil precursors carrying long carbon chains in the 3,4,5-positions were found to be difficult. In fact, the yield of benzil formation strongly depends on (i) the position, (ii) the nature and (iii) on the number of substituents on the starting benzaldehyde. Thus, in order to introduce 12 carbon chains around the nickel-bis(dithiolene) core and to eventually obtain room-temperature liquid crystalline materials, another strategy was explored here and gallate derivatives, carrying long carbon chains in the 3,4,5-positions, were grafted on preformed benzil precursors through ester or amide bonds.
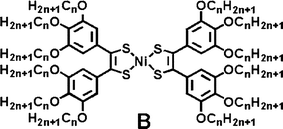
In the present work, we report the design and the successful synthesis of new benzil protomesomorphic ligands substituted at both ends by gallate derivatives through ester or amide linkers, able to generate, after metal complexation, room-temperature liquid crystalline metal-bis(dithiolene) complexes. Their electronic properties have been determined by optical and electrochemical investigations. The thermotropic liquid crystalline properties of the functional benzyl derivatives and their corresponding nickel complexes have been investigated: all nickel-bis(dithiolene) complexes are mesomorphic, forming columnar mesophases over wide thermal ranges. An original cubic mesophase was even observed for one of them. The importance of the linker (ester, amide) on the thermal behavior of both ligands and complexes will be discussed and packing models in mesophases proposed.
Experimental
300.1 (1H) and 75.5 MHz (13C) NMR spectra were recorded on a Bruker Avance 300 spectrometer at room temperature using perdeuterated solvents as internal standards. FT-IR spectra were recorded using a Varian-640 FT-IR spectrometer. UV-Vis-NIR spectra were recorded using a Cary 5000 UV-Vis-NIR spectrophotometer (Varian, Australia). Elemental analysis were performed at the Centre Régional de Mesures Physiques de l′Ouest, Rennes. Cyclic voltammetry were carried out on a 10−3 M solution of complex in CH2Cl2, containing 0.2 M nBu4NPF6 as supporting electrolyte. Voltammograms were recorded using an Autolab electrochemical analyser (PGSTAT 30, Ecochemie BV). The reference electrode was SCE and the counter electrode was graphite. Polarising optical microscopy images were taken with a Nikon H600L polarising microscope. Differential scanning calorimetry (DSC) was carried out by using a NETZSCH DSC 200 F3 instrument equipped with an intracooler. DSC traces were measured at 10 °C min−1 down to −30 °C. The XRD patterns were obtained with a transmission Debye–Scherrer-like geometry. A linear monochromatic Cu-Kα1 beam (λ = 1.5405 Å) was obtained using a sealed-tube generator (900 W) equipped with a bent quartz monochromator. The crude powder was filled in Lindemann capillaries of 1 mm diameter and 10 μm wall-thickness. In each case, exposure times were varied from 1 to 4 h. The diffraction patterns were recorded with a curved Inel CPS120 counter gas-filled detector linked to a data acquisition computer (periodicities up to 60 Å) and on image plates scanned by STORM 820 from Molecular Dynamics with 50 μm resolution (periodicities up to 100 Å); the sample temperature was controlled within ±0.05 °C in the 20–200 °C temperature range.4-Methoxybenzaldehyde (99%) was purchased from Sigma Aldrich and used as received. NiCl2 (99.9%), was purchased from Alfa Aesar and used without further purification. DMI (99%), P4S10 (98%) and hydrobromic acid (48%) were purchased from Acros Organics. Anhydrous CH2Cl2 (Sigma-Aldrich) and triethylamine (Alfa Aesar) were obtained by distillation over P2O5 and KOH respectively. The reactions were followed using thin layer chromatography (TLC) plates revealed by a UV-lamp at 254 nm. All other reagents and materials from commercial sources were used without further purification. Silica gel used in chromatographic separations was obtained from Acros Organics (Silica Gel, ultra pure, 40–60 μm). 4,4′-Dihydroxybenzil,18 4,4′-diaminobenzil19 and 3,4,5-trialkoxybenzoic acids20 were prepared as previously described in the literature. Full synthetic details and characterizations of the Cnbenzilester and Cnbenzilamide compounds (n = 8, 12, 16) and their corresponding CnesterNi and CnamideNi nickel complexes are given in ESI†.
Results and discussion
Synthesis and characterisation
The synthetic route for the preparation of the CnesterNi and CnamideNi complexes with n = 8, 12, 16 is shown in Scheme 1. Starting 4,4′-dihydroxybenzil18 and 4,4′-diaminobenzil19 compounds were synthesized according to reported procedures. Cnbenzilester and Cnbenzilamide compounds with n = 8, 12 and 16 were readily obtained by reaction of the 4,4′-dihydroxybenzil or 4,4′-diaminobenzil with 3,4,5-trialkyloxybenzoic acid chloride derivatives20 (n = 8, 12, 16) under anhydrous conditions using triethylamine as base (Scheme 1). Purification of the compounds was achieved by chromatography on silica gel followed by recrystallization from CH2Cl2–MeOH. The ester and amide ligands were isolated in good yield as light-yellow powders (C8benzilester, 55%; C12benzilester, 43%; C16benzilester, 60%; C8benzilamide, 51%; C12benzilamide, 48%; C16benzilamide, 59%). 1H NMR spectra obtained in CDCl3 of the Cnbenzilester compounds display a singlet peak in the aromatic region (ca. 7.4 ppm), characteristic of the aromatic protons on the gallate substituents and an AB system at 7.37–7.38 and 8.09–8.10 ppm which is attributed to the protons localized on the benzil fragment. Formation of the ester bond is also confirmed by the presence of a characteristic band at 1735 cm−1 on the infrared spectra. As for the amide compounds, in addition to the singlet peak (7.04–7.06 ppm) and AB system (7.81–7.82 and 8.03–8.04 ppm) assigned to the gallate and benzil protons respectively, another singlet was observed at 7.88–7.91 ppm, corresponding to the NH proton. The formation of the amide function was also confirmed by IR spectroscopy with the presence of a characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signal at 1640–1680 cm−1.
O signal at 1640–1680 cm−1.
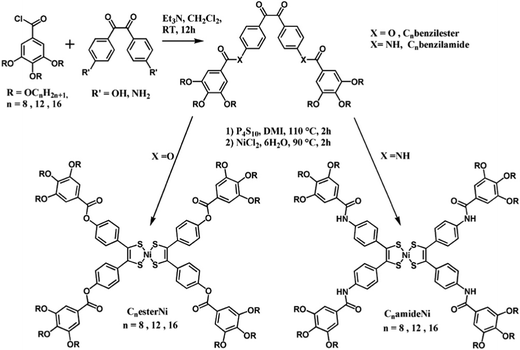 | ||
| Scheme 1 Syntheses of the Cnbenzilester and Cnbenzilamide ligands and corresponding nickel-bis(dithiolene) complexes (R = OCnH2n+1 with n = 8, 12 and 16). | ||
The preparation of phenyl-substituted nickel dithiolene complexes is most often based on sulfiding of the benzil moieties with P4S10, followed by the hydrolysis of the intermediate phosphorous thioester derivatives in the presence of a nickel salt such as NiCl2·6H2O to directly afford the oxidized, neutral nickel complex. This strategy was successfully used here for the preparation of the CnesterNi and CnamideNi complexes with n = 8, 12, 16, from the corresponding benzilester and benzilamide ligands. 1,3-Dimethyl-2-imidazolidinone (DMI) was used as solvent rather than dioxane, to improve the yield of the syntheses.21 After purification by chromatography on silica gel followed by crystallization from CH2Cl2–MeOH, the complexes were isolated in good yields as dark-green powders (C8esterNi, 34%; C12esterNi, 58%; C16esterNi, 44%; C8amideNi, 44%; C12amideNi, 46%; C16amideNi, 42%). The purity of the ester complexes was unambiguously confirmed by 1H and 13C NMR spectroscopy, IR spectroscopy and elemental analysis. After sulfuration and metal complexation, the doublets from the AB system, characteristic of the benzil fragment, shift upfield from 7.37–7.38 and 8.09–8.10 ppm to 7.17–7.20 and 7.50–7.52 ppm. The complex formation is also ascertained by the complete disappearance of the characteristic vibration band of the benzil function (Ph–CO–CO–Ph) localized at 1672 cm−1 on the IR spectra of the Cnbenzilester compounds. In contrast, no clear NMR spectra were obtained for the amide complexes, likely due to a strong tendency of the complexes to aggregate in solution and in the solid state through hydrogen bonding between the amide fragments.22 Nevertheless, the appearance of a characteristic dark green colour of the nickel-bis(dithiolene) complexes, the disappearance of the peak characteristic of the benzil function (Ph–CO–CO–Ph) located at 1654 cm−1 in the IR spectrum and the elemental analysis confirmed the formation of the CnamideNi complexes with n = 8, 12, 16.
Electronic properties
Optical and electrochemical data for the CnesterNi and CnamideNi complexes (n = 8, 12, 16) determined in solution are given in Table 1. The absorption spectra of CnesterNi complexes (n = 8, 12, 16) show a main absorption band peak at 284 nm with a shoulder at 308 nm (Fig. 1 and Table 1) in the UV region. These absorption bands are assigned to π–π* transitions localized on the dithiolene ligands. Another strong absorption band centred at 866 nm, extending from 700 nm up to 1100 nm with high extinction coefficients (ε ≈ 30000 M−1 cm−1), is also observed in the near-IR region and is characteristic of the neutral Ni-bis(dithiolene) complexes. This low-energy absorption band is attributed to an electronic transition from the HOMO (Lπ) of b1u symmetry to the LUMO (Lπ*–αdxy) with a metallic character of b2g symmetry.12b Similarly, CnamideNi complexes (n = 8, 12, 16) display a strong absorption band in the near-IR region centred at 920 nm, extending from 750 nm up to 1100 nm. By comparison with complexes A described by Ohta,12 the near-IR absorption band is centred at even lower energy, 933 nm (Table 1).21b The observed hypsochromic shift in the CnesterNi and CnamideNi complexes (n = 8, 12, 16) can be understood by the electron-withdrawing ability of the various connecting functions used here, that increases on going from the ether (molecules A) to amide linkage and from amide to ester linkage. It is noted that there is no influence of the length of the carbon chains on the electronic properties of the Ni complexes.
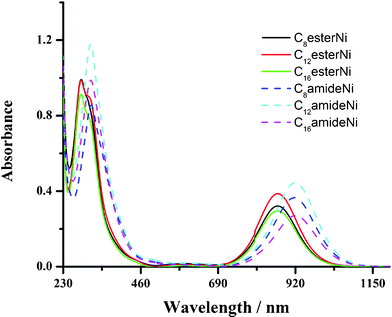 | ||
| Fig. 1 (a) UV-vis-NIR spectra of CnesterNi and CnamideNi complexes (n = 8, 12, 16) in dichloromethane (c ∼ 10−5 mol l−1). | ||
| Complex | λ a/nm | ε a/M−1 cm−1 | E 1/2(−1/−2)b/V | E 1/2(0/−1)b/V | E 1/2(+1/0)b/V |
|---|---|---|---|---|---|
| a Determined in dichloromethane solution (c ∼ 10−5 M). b In 0.1 M nBu4NPF6–dichloromethane at room temperature; scan rate, 100 mV s−1; E1/2 = (Epa + Epc)/2; Epa and Epc are the anodic peak and the cathodic peak potentials, respectively. c Measured at 50 °C. | |||||
| C8esterNi | 282 | 99300 |
−0.70 | 0.16 | — |
| 867 | 32200 |
||||
| C12esterNi | 283 | 98700 |
−0.68 | 0.17 | — |
| 867 | 38700 |
||||
| C16esterNi | 282 | 91400 |
−0.74c | 0.07c | — |
| 867 | 29600 |
||||
| C8amideNi | 313 | 85600 |
−0.78 | 0.02 | — |
| 917 | 36500 |
||||
| C12amideNi | 311 | 117800 |
−0.79 | 0.0 | — |
| 919 | 44500 |
||||
| C16amideNi | 312 | 98900 |
−0.8c | −0.01c | — |
| 919 | 27300 |
||||
| Molecule A (n = 12)21b | 933 | 31300 |
−1.33 | −0.10 | 0.915 |
Electrochemical properties of nickel complexes were investigated by cyclic voltammetry in CH2Cl2. The CnesterNi complexes (n = 8, 12, 16) exhibit two reversible reduction processes centred at 0.1 and −0.7 V (vs. SCE), corresponding to the formation of the monoanionic and the dianionic species, respectively (Fig. 2 and Table 1). The redox potentials are weakly affected by the variation of the length of the hydrocarbon chain. Comparison with molecule A reveals an anodic shift of the redox potentials which, again, is due to the electron-withdrawing ability of the ester linkage. Introduction of four electron-withdrawing ester linkages renders the reduction of the complex more facile than that of complex A with ether linkages. Cyclic voltammograms of CnamideNi complexes (n = 8, 12, 16) also display two reversible reduction processes centred at 0.0 V (0 → −1) and −0.8 V (−1 → −2) (vs. SCE). The reduction potentials are slightly more cathodic than for the corresponding CnesterNi complexes, a behaviour in line with a stronger electron-withdrawing ability of the ester linkage. As for the ester complexes, there is no real influence of the carbon chain length.
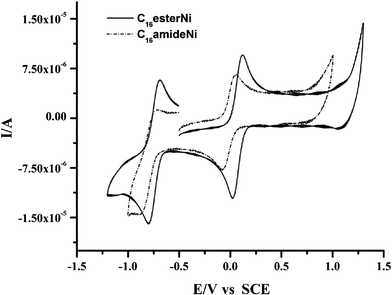 | ||
| Fig. 2 Cyclic voltammograms of C16esterNi and C16amideNi in dichloromethane (vs. SCE, v = 100 mV s−1, electrolyte 0.2 M nBu4NPF6). | ||
Thermal studies
The thermal behavior of the Cnbenzilester and Cnbenzilamide proligands (n = 8, 12 and 16) and of their corresponding nickel complexes was investigated by a combination of polarizing optical microscopy (POM), differential scanning calorimetry (DSC) and small-angle X-ray diffraction (SA-XRD). The measured transition temperatures, enthalpy values and mesophase parameters are given in Table S1 in ESI†.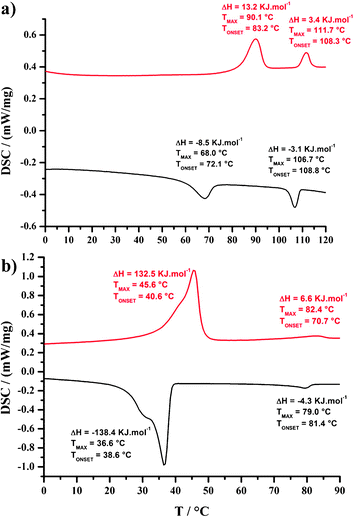 | ||
| Fig. 3 DSC traces of (a) C12benzilamide and (b) C16esterNi (top: second heating curve; bottom: first cooling curve). | ||
 | ||
| Fig. 4 (a) C12benzilamide and (b) C12esterNi complex observed by optical microscopy between crossed-polarizers at 105 and 115 °C, respectively (crossed-polarizers symbolized by the white cross in the corner of the picture). | ||
Finally for the C12benzilamide compound, no clear texture change was observed at 90 °C, and XRD confirmed the existence of a low-temperature Colr phase down to room temperature (no phase transition down to 0 °C) (Fig. 5a). The pattern exhibits a series of four sharp, small-angle peaks that can either be indexed as (11), (20), (22), (42) or as the (20), (11), (40), (60) reflections of a rectangular lattice with c2mm plane group (both solutions are equiprobable and compatible with the centred lattice, Table S1, ESI†).24 These peaks are also associated with a broad signal at 4.6 Å (hch) confirming the molten state of the alkyl chains as found for the high-temperature Colh.
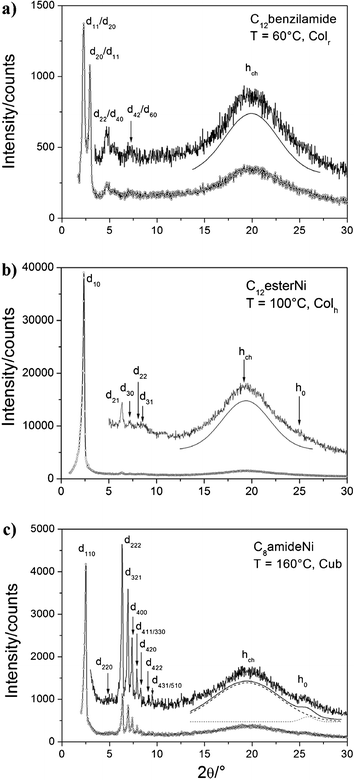 | ||
| Fig. 5 XRD Patterns of C12benzilamide at 60 °C in the Colr phase, C12esterNi at 100 °C in the Colh phase and C8amideNi at 160 °C in the cubic phase respectively. | ||
:√7:3, were respectively indexed as the (10), (21) and (30) reflections of a hexagonal lattice, with a parameter of 38.8 Å (Fig. S10 and Table S1, ESI†). The large number of small-angle reflections observed in the lower temperature mesophase (Fig. S11 and Table S1, ESI†), consistent with a substantial symmetry breaking (e.g. reflections h + k = 2n + 1), is typical for a pseudo-hexagonal mesophase with non-centered rectangular p2gg lattice.25 In the wide angle region, a broad halo centred at 4.5 Å (hch) confirms the liquid crystalline nature of the phases. Two additional signals (thereafter labelled hM and h0) centred at 4.05 and 3.55 Å, respectively were also observed in the wide-angle region. The low intensity broad peak at 3.55 Å, which is classically observed in the few reported metal-bis(dithiolene) mesophases,11,12 is attributed to the stacking distance between the NiS4 cores (h0). The small peak at 4.05 Å (hM) is likely attributed to some residual interactions between the benzyl-ester arms, which are also probably slightly rotated from the Ni-S4 mean plane for steric constraints.
The DSC traces of the C12esterNi complex displays three reversible transitions located at −10, 90 and 125 °C (Fig. S12, ESI†). The broad transition observed at −10 °C is attributed to the crystallization, whereas above 125 °C, the compound is isotropic. Between −10 and 125 °C, texture characteristic of columnar mesophases were observed (Fig. 4b). No clear texture change was observed around 90 °C. XRD confirmed the existence of two mesophases, unambiguously characterized as Colh and Colr phases; the hexagonal symmetry was deduced for the upper phase from the presence of five sharp small-angle reflections in the expected ratio (Fig. 5b), and the rectangular symmetry p2gg (Fig. S13, ESI†) was evidenced by the presence of additional peaks, consistent with the re-organization of the local packing of the columns, as C8esterNi.
Finally, the C16esterNi complex displays a broad and energetic thermal transition at 46 °C and a second weak and broad transition centered at 82 °C (Fig. 3b). The low-temperature transition is attributed to a crystal-to-mesophase transition whereas the high-temperature transition corresponds to the mesophase-to-isotropic liquid transition. In this temperature interval, a fluid texture characteristic of columnar phase is observed, the hexagonal nature of which was confirmed by XRD (Table S1 and Fig. S14, ESI†).
These results highlight that complexation of a metal center by two non-mesogenic ester ligands leads to the formation of a mesogenic molecules that can form one (n = 16), and even two (n = 8, 12) stable columnar mesophases. Mesophase stability is increased as the pendant aliphatic chain-lengths are decreased and, interestingly, room-temperature columnar mesophases containing redox-active metal-bisdithiolene cores are isolated here for the first time. Now, one can wonder what will be the effect of the introduction of an amide linker on the thermotropic properties of the nickel-bisdithiolene complexes obtained after metal complexation by the mesogenic benzil Cnbenzilamide proligands?
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.26 Along with these reflections, an intense halo corresponding to the molten state of the chains (hch = 4.5 Å) and a weak halo (more or less visible) around 3.5 Å (h0) corresponding to short-range π–π stacking interactions (Fig. 5c, Table S1, ESI†) were also observed.
m space group.26 Along with these reflections, an intense halo corresponding to the molten state of the chains (hch = 4.5 Å) and a weak halo (more or less visible) around 3.5 Å (h0) corresponding to short-range π–π stacking interactions (Fig. 5c, Table S1, ESI†) were also observed.
Finally, both higher homologs C12amideNi and C16amideNi display a broad and reversible transition at −18 and 30 °C, respectively, attributed to the melting of a crystalline phase (Fig. S17 and S19, ESI†) and a second weak and reversible transition at 177 and 115 °C, respectively, corresponding to the isotropisation of the compounds. Between these temperature intervals, XRD patterns confirm the formation of a columnar hexagonal phase. For C12amideNi, the five sharp peaks in the low-angle region can be indexed in a 2D hexagonal lattice (a = 42.7 Å, Table S1, Fig. S18, ESI†). For C16amideNi, only one peak has been detected in the small-angle region on the XRD patterns measured between 30 and 115 °C (Fig. S20, ESI†) but on the basis of the POM observations, and thanks to optical textures reminiscent of hexagonal phase, the mesophase can nevertheless be safely assigned as Colh.
The thermal behaviours of the CnesterNi and CnamideNi complexes with n = 8, 12, 16 are summarized in Table 2 and in Fig. 6 (see ESI† for full details). All the complexes remain in liquid-crystalline states over wide thermal ranges, especially at room temperature (except C16 complexes), with a constant decrease of the clearing temperature as the chain length is increased.27 Such room-temperature columnar mesophases, containing metal-bisdithiolene cores, potentially able to efficiently carry charges when stacked into columnar structures, can be of great interest for applications in electronic devices using room-temperature liquid crystalline properties. It can also be noticed that the introduction of amide functions leads to a stronger stabilization of the hexagonal columnar phase with a decrease of the melting points and an increase of the isotropization points, when compared to the CnesterNi ester compounds, likely due to the formation of additional hydrogen bonding networks (vide infra). At room temperature, CnesterNi compounds allow isolation of rectangular columnar phases whereas CnamideNi compounds form hexagonal columnar phases. It is interesting to note that C12amideNi compound is in a hexagonal columnar mesophase over a temperature range of 195 °C from −18 up to 177 °C. In the series of CnamideNi complexes, a original cubic phase is observed at high temperature above the Colh phase.
| Compound | State T/°C (ΔH/kJ mol−1) | Mesophase parameters |
|---|---|---|
| a Cr, crystalline phase; Iso, isotropic liquid; Colh, hexagonal columnar mesophase; Colr, rectangular columnar mesophase; Cub, cubic phase; decomp. = decomposition temperature (evaluated by POM observations); a and b are the lattice parameters of the mesophases; Sh and Sr are the columnar cross-section area of hexagonal and rectangular phases, and Vcub, the volume of the cubic lattice, respectively; Z is the number of complexes per columnar slice of thickness h (h = h0); Nagg is the number of complexes within the cubic lattice. See ESI for full details. b Broad transition. | ||
| C8esterNi | Heating: Colr 88.5b Colh 142 (10.0) Iso | Colh-p6mm (T = 120 °C) |
| Cooling: Iso 139 (−9.7) Colh 87b Colr | a = 38.8 Å, Sh = 1305 Å2, Z ∼ 1 | |
| Colr-p2gg (T = 80 °C) | ||
| a = 68.42 Å, b = 39.54 Å | ||
| S r = 1351 Å2, Z ∼ 1.1 | ||
| C12esterNi | Heating: Cr −10b Colr 90 (10.3) Colh 125 (9.7) Iso | Colh-p6mm (T = 100 °C) |
| Cooling: Iso 120 (−8.4) Colh 86.5 (−11.0) Colr −15b Cr | a = 42.67 Å, Sh = 1577 Å2, Z ∼ 1 | |
| Colr-p2gg (T = 40 °C) | ||
| a = 74.8 Å, b = 43.2 Å | ||
| S h = 1615 Å2, Z ∼ 1.05 | ||
| C16esterNi | Heating: Cr 46 (132.5) Colh 82 (6.6) Iso | Colh-p6mm (T = 60 °C) |
| Cooling: Iso 79 (−4.3) Colh 37 (−138.4) Cr | a = 46.36 Å, Sh = 1861 Å2, Z ∼ 1 | |
| C8amideNi | Heating: Colh 156 (10.0) Cub 230 (decomp.) | Colh-p6mm (T = 100 °C) |
| Cooling: Cub 140 (−8.2) Colh | a = 38.7 Å, Sh = 1295.8 Å2, Z ∼ 1 | |
| Cub-Imm (T = 160 °C) |
||
| a = 47.3 Å, Vcub ∼ 106 Å3, Nagg ∼ 20–24 | ||
| C12amideNi | Heating: Cr −18b Colh 177 (4.5) Iso | Colh-p6mm (T = 100 °C) |
| Cooling: Iso 155 (−4.8) Colh −20b Cr | a = 42.7 Å, Sh = 1580.8 Å2, Z ∼ 1 | |
| C16amideNi | Heating: Cr 30 (155.4) Colh 115 (3.1) Iso | Colh-p6mm (T = 50 °C) |
| Cooling: Iso 82 (−2.3) Colh 23 (−149.2) Cr | a = 48.0 Å, Sh = 1998.5 Å2, Z ∼ 1 | |
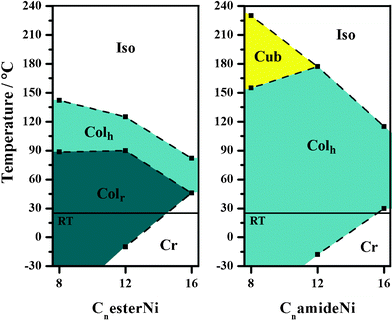 | ||
| Fig. 6 Thermal behaviour of CnesterNi and CnamideNi complexes as a function of chain length (n = 8, 12, 16) (room temperature (RT) = 25 °C). | ||
Infrared spectra of the mesophases
The strong stabilization of the columnar phases observed with the amide compounds, when compared with the ester ones, is certainly induced by the formation of an intermolecular hydrogen bonding network. To address this point, the amide functions were studied by FT-IR spectroscopy, a powerful tool to probe hydrogen bonding and ordering of hydrocarbon chains. FTIR measurements performed in the room-temperature Colh or Colr mesophases of the precursor Cnbenzilamide (n = 8, 12) compounds as well as on the CnamideNi (n = 8, 12) complexes at 25 °C, after heating at 118, 120, 160 and 180 °C respectively, confirm that the molecular organization is stabilized by hydrogen-bonding interactions, as clearly evidenced by the νNH and νCO stretching vibrations that lie respectively at 3282–3300 and 1652–1653 cm−1. Note that corresponding values for the free amides are at 3400 cm−1 for νNH and around 1680 cm−1 for νCO.28 The presence of single νCO and νNH stretching vibrations on the FTIR spectra also indicate the formation of a hydrogen-bonded network involving all amide functions. The frequencies of the bands due to CH2 antisymmetric and symmetric modes [νa(CH2) and νs(CH2)] of the alkyl chains appear at ca. 2920–2924 and ca. 2851–2853 cm−1 in the mesophase and also indicate that the hydrocarbon chains are all in trans conformation.29 These trans peaks shift indeed to ≈2926 and 2855 cm−1 respectively, if the population of gauche form in the alkyl chains increases.30Packing study of the liquid crystalline phases
The mesomorphism of all the mesogens described here is essentially characterized by the formation of columnar mesophases, and therefore the molecules are likely to stack on top of each other in order to generate columns with circular cross-sections (as in Colh phase) or non-circular cross-sections (reduced symmetry as in Colr phase).24 The columnar packing is characterized by the columnar cross-section, Scol, and the stacking periodicity h along the columnar axis, both parameters being analytically linked through the relation h × Scol = Z × VM, where Z is the number of molecules within a columnar stratum (disc) h-thick and VM the molecular volume. With the Cnbenzilamide precursors, the formation of columns requires the association of at least two mesogenic molecules, likely in a side-by-side type as classical hexacatenar materials,24 to generate a “supramolecular” discotic slice h-thick that stacks into columns (Fig. 7, left). Note here that benzil molecules can adopt either a transoid or cisoid conformation: in solution, the transoid form must be the predominant one in order to minimize dipolar interactions, whereas in columnar mesophases, pairing of the cisoid forms will allow a better space filling of the discs and the introduction of stabilizing dipolar interactions. The absence of short-range periodicities on the XRD patterns of the benzilamide precursors, except the carbon chain interactions, indicates indeed that there is no interaction between the benzil cores and that the columns are generated by a continuum of molecules in a fluid state, and consequently the average thickness of such a supramolecular aggregate is given by hch. In fact, aggregation may take place in such a way that the molecules contained in a “slice” are not in the same plane, but just point their tips towards the column core without the need to form discrete discs. On the other hand, in the case of the nickel complexes, due to their disc-like shape, only one mesogen is necessary to form a columnar slice (Fig. 7, right). The reduction of the phase symmetry of the planar arrangements (from hexagonal to pseudo-hexagonal) of the columns results from the combination of a more or less pronounced tilt of the molecular plane with respect to the lattice plane. The tilt, which increases with temperature decrease, concomitantly hinders rotation about the columnar axis, and freezes the different orientations (random orientations) of the elliptical projection of the columnar cross sections onto the lattice plane. The presence of a stacking distance h0 with the Ni complexes, although very weak and partially hidden by the strong scattering of the alkyl chains, clearly indicates some short-range stacking of the extended cores. The columns are generated by the stacking of the discs and the mutual organisation of these columns into a hexagonal or rectangular lattice leads to the formation of the mesophase. An alternated π/2 stacking along the columnar axis likely occurs between two consecutive slices to optimize molecular packing within the mesophase (Fig. 7), but obviously is not correlated over too long distances.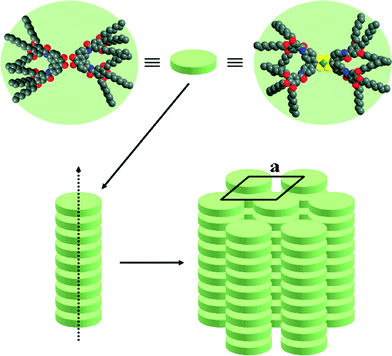 | ||
| Fig. 7 Suggested molecular organisation of the Cnbenzilamide (n = 8, 12, 16) precursors (two molecules per plateau) (left) and of the nickel complexes CnesterNi or CnamideNi (one molecule per plateau) (right). | ||
Thermotropic cubic phases, as ordered three-dimensional supramolecular edifices, with body-centered space groups are not so common in thermotropic liquid crystals,17,31 and their structure is not well understood yet. It may consist, either of bicontinuous, interwoven rod-networks32 or alternatively, of a discontinuous three-dimensional arrangement of pseudo-spherical micelles located at the nodes of the cubic lattice.33 The formation of a bicontinuous cubic structure seems more reasonable since it appears difficult to arrange such discoid complexes into micelles. The generation of a bicontinuous cubic structure would thus result from the perturbation at short range of the stacking, consequent to substantial molecular conformational changes, forcing the molecules to develop interactions in the three spatial directions.
Conclusion
Following the original dialkoxy derivatives described more than twenty years ago, we have identified here two new families of protomesogenic ligands with added ester or amide functionalities. After sulfuration and metal complexation of these rationally designed benzil proligands, original functional metallomesogens with a nickel bis(dithiolene) core have been isolated. They all exhibit columnar phases over large temperature ranges especially around room temperature. CnesterNi complexes form columnar mesophases of rectangular and hexagonal symmetry whereas the amide linkage in CnamideNi complexes strongly stabilize hexagonal columnar mesophases down to room temperature, as illustrated by C12amideNi which display a Colh mesophase from −18 up to + 177 °C. The role of hydrogen bonding between the amide functions in the stabilisation of the mesophases was confirmed by IR spectroscopy. Furthermore, with the C8amideNi compound, a cubic phase, stable over ∼50 °C, from 155 to 203 °C, and never observed up to now with metal-bis(dithiolene), was clearly identified. The most probable model suggests that the short-range molecular stacks would perturb the long-range 2D supramolecular arrangement, and generate instead the formation of two distinct, interdigitated cylindrical 3D networks.The strategy adopted here has allowed for the formation of columnar mesophases of hexagonal or rectangular symmetry at room temperature. Such materials, also strongly absorbing in the NIR region, can be of potential interest, as active layers, in optoelectronic applications. Future work will be first devoted to the measurements of charge motilities in these new systems.
Acknowledgements
Financial support from the Region Bretagne (Post doctoral grant to S. D.) and from the University Rennes 1 (Incitative Action 2010) is gratefully acknowledged. BD thanks the CNRS-Université de Strasbourg for support and Dr Benoît Heinrich for fruitful discussions.References
- J.-M. Lehn, Supramolecular Chemistry, Wiley-VCH, New-York 1995 Search PubMed.
- (a) G. K. Such, A. P. R. Johnston and F. Caruso, Chem. Soc. Rev., 2011, 40, 19–29 RSC; (b) S. Yamamichi, Y. Jinno, N. Haraya, T. Oyoshi, H. Tomitori, K. Kashiwagi and M. Yamanaka, Chem. Commun., 2011, 47, 10344–10-346 RSC; (c) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS; (d) T. Kato, Science, 2002, 295, 2414–2418 CrossRef CAS; (e) I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS.
- (a) M. Grell and D. D. C. Bradley, Adv. Mater., 1999, 11, 895–905 CrossRef CAS; (b) Y. Shimizu, K. Oikawa, K.-I. Nakayama and D. Guillon, J. Mater. Chem., 2007, 17, 4223–4229 RSC; (c) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS.
- (a) S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS; (b) S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC; (c) S. Kumar, Chem. Soc. Rev., 2006, 35, 83–109 RSC; (d) W. Pisula, M. Zorn, J. Young, C. K. Müllen and R. Zentel, Macromol. Rapid Commun., 2009, 30, 1179–1202 CrossRef; (e) B. R. Kaafarani, Chem. Mater., 2011, 23, 378–396 CrossRef CAS; (f) S. Kumar, Liq. Cryst., 2009, 36, 607–638 CrossRef CAS.
- (a) A. M. van de Craats, N. Stutzmann, O. Bunk, N. N. Nielsen, M. Watson, K. Müllen, H. D. Chanzy, H. Sirringhaus and R. H. Friend, Adv. Mater., 2003, 15, 495–499 CrossRef CAS; (b) W. Pisula, A. Menon, M. Stepputat, I. Lieberwirth, U. Kolb, A. Tracz, H. Sirringhaus, T. Pakula and K. Müllen, Adv. Mater., 2005, 17, 684–689 CrossRef CAS; (c) T. Kitamura, S. Nakaso, N. Mizoshita, Y. Tochigi, T. Shimomura, M. Moriyama, K. Ito and T. Kato, J. Am. Chem. Soc., 2005, 127, 14769–14775 CrossRef CAS; (d) J.-P. Hong, M.-C. Um, S.-R. Nam, J.-I. Hong and S. Lee, Chem. Commun., 2009, 310–312 RSC.
- (a) U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC; (b) T. Hassheider, S. A. Benning, H.-S. Kitzerow, M.-F. Achard and H. Bock, Angew. Chem., Int. Ed., 2001, 40, 2060–2063 CrossRef CAS.
- (a) M. A. Loi, E. Da Como, R. Zamboni and M. Muccini, Synth. Met., 2003, 139, 687–690 CrossRef CAS; (b) M. A. Loi, E. Da Como, F. Dinelli, M. Murgia, R. Zamboni, F. Biscarini and M. Muccini, Nat. Mater., 2004, 4, 81–85 Search PubMed.
- (a) B. A. Greeg, M. A. Fox and A. J. Bard, J. Phys. Chem., 1990, 94, 1586 CrossRef CAS; (b) A. M. Fox, J. V. Grant, D. Melamed, T. Torimoto, C.-Y. Liu and A. J. Bard, Chem. Mater., 1998, 10, 1771 CrossRef CAS; (c) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS; (d) W. Kubo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374–4381 CrossRef CAS.
- (a) B. Donnio, D. Guillon, D. W. Bruce and R. Deschenaux, Metallomesogens, in Comprehensive Coordination Chemistry II: From Biology to Nanotechnology, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, UK 2003, vol. 7 (ed. M. Fujita and A. Powell), ch. 7.9, pp. 357–627 Search PubMed; (b) R. W. Date, E. Fernandez Iglesias, K. E. Rowe, J. M. Elliott and D. W. Bruce, Dalton Trans., 2003, 1914–1931 RSC.
- (a) M. C. Aversa, P. Bonaccorsi, D. W. Bruce, F. Caruso, P. Giannetto, S. Lanza and S. Morrone, Inorg. Chim. Acta, 1997, 256, 235–2415 Search PubMed; (b) M. C. Aversa, P. Bonaccorsi, D. W. Bruce, F. Caruso, B. Donnio, P. Giannetto, D. Guillon, S. Lanza and S. Morrone, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2000, 348, 53–64 Search PubMed.
- (a) K. Ohta, H. Ema, I. Yamamoto and K. Matsuzaki, Liq. Cryst., 1988, 3, 1671–1687 CAS; (b) K. Ohta, Y. Morizumi, H. Ema, T. Fujimoto and I. Yamamoto, Mol. Cryst. Liq. Cryst., 1991, 208, 55–63 CAS.
- (a) K. Ohta, H. Hasebe, H. Ema, T. Fujimoto and I. Yamamoto, J. Chem. Soc., Chem. Commun., 1989, 1610–1611 RSC; (b) K. Ohta, H. Hasebe, H. Ema, M. Moriya, T. Fujimoto and I. Yamamoto, Mol. Cryst. Liq. Cryst., 1991, 208, 21–32 CrossRef CAS; (c) K. Ohta, H. Hasebe, M. Moriya, T. Fujimoto and I. Yamamoto, Mol. Cryst. Liq. Cryst., 1991, 208, 33–41 CrossRef CAS; (d) K. Ohta, Y. Inagaki-Oka, H. Hasebe and I. Yamamoto, Polyhedron, 2000, 19, 267–274 CrossRef CAS.
- R. Perochon, C. Poriel, O. Jeannin, L. Piekara-Sady and M. Fourmigué, Eur. J. Inorg. Chem., 2009, 5413–5421 CrossRef CAS.
- J.-Y. Cho, B. Domercq, S. C. Jones, J. Yu, X. Zhang, Z. An, M. Bishop, S. Barlow, S. R. Marder and B. Kippelen, J. Mater. Chem., 2007, 17, 2642–2647 RSC.
- R. Perochon, P. Davidson, S. Rouzière, F. Camerel, L. Piekara-Sady, T. Guizouarn and M. Fourmigué, J. Mater. Chem., 2011, 21, 1416–1422 RSC.
- T.-T. Bui, O. Thiebaut, E. Grelet, M.-F. Achard, B. Garreau-de Bonneval and K. I. Moineau-Chane Ching, Eur. J. Inorg. Chem., 2011, 2663–2676 Search PubMed.
- A. Escande, L. Guénée, H. Nozary, G. Bernardinelli, F. Gumy, A. Aebischer, G. Bünzli J.-C, B. Donnio, D. Guillon and C. Piguet, Chem.–Eur. J., 2007, 13, 8696–8713 CrossRef.
- H. Horie, A. Takagi, H. Hasebe, T. Ozawa and K. Otha, J. Mater. Chem., 2001, 11, 1063–1071 RSC.
- H. L. Gee and J. Harley-Mason, J. Chem. Soc., 1947, 251 Search PubMed.
- F. Camerel, G. Ulrich and R. Ziessel, Org. Lett., 2004, 6, 4171–4174 CrossRef CAS , and references therein.
- (a) K. Takuma, Y. Irizato and K. Katho, PCT Int. Appl., 1990, 22, WO9012019 Search PubMed; (b) R. Perochon, L. Piekara-Sady, W. Jurga, R. Clérac and M. Fourmigué, Dalton Trans., 2009, 3052–3061 RSC.
- F. Camerel, B. Donnio, C. Bourgogne, M. Schmutz, D. Guillon, P. Davidson and R. Ziessel, Chem.–Eur. J., 2006, 12, 4261–4274 CrossRef CAS.
- (a) J. Malthête, H. T. Nguyen and C. Destrade, Liq. Cryst., 1993, 13, 171–187 CrossRef CAS; (b) H. T. Nguyen, C. Destrade and J. Malthête, Adv. Mater., 1997, 9, 375–388 CrossRef CAS; (c) D. Fazio, C. Mongin, B. Donnio, Y. Galerne, D. Guillon and D. W. Bruce, J. Mater. Chem., 2001, 11, 2852–2863 RSC.
- (a) B. Donnio, B. Heinrich, H. Allouchi, J. Kain, S. Diele, D. Guillon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 15258–15268 CrossRef CAS; (b) F. Morale, R. W. Date, D. Guillon, D. W. Bruce, R. L. Finn, C. Wilson, A. J. Blake, M. Schröder and B. Donnio, Chem.–Eur. J., 2003, 9, 2484–2501 CrossRef CAS; (c) J. Matraszek, J. Mieczkowski, D. Pociecha, E. Gorecka, B. Donnio and D. Guillon, Chem.–Eur. J., 2007, 13, 3377–3385 CrossRef CAS.
- The rectangular lattice is primitive (for example p2gg), as shown by the (12) which excludes centered groups according the extinction rule h + k = 2n + 1. Actually, the phase structure likely results from a symmetry breaking of the local hexagonal packing of columns, leading to a doubling of the lattice periodicities with parameters a and b of 57.92 and 33.44 Å, respectively (a/b = √3). The symmetry break can result from a shift between the columns of intercalated row from the lattice center.
- The reflections, in the ratio √2:√8:√12:√14:√16:√18:√20:√24:√26, are all theoretically compatible with the reflection conditions of cubic space groups with simple (cs) or body-centred (bcc) symmetry, but totally excludes the cfc system (F) [reflection conditions, 0kl: k, l = 2n (and k + l = 4n), hhl: h + l = 2n or h, l = 2n, h00: h = 2n or 4n]. The absence of numerous low-angle reflections, not only explained by the groups' extinction conditions, also exclude the cs groups with symmetry Pm, Pn, Pmm, Pmn, Pnn and Pnm. The reflections were thus indexed as (110), (220), (222), (321), (400), (330/411), (420), (422), and (431/510) of the (bcc) groups Im and Imm [reflection conditions, 0kl: k + l = 2n, hhl: l = 2n, h00: h = 2n] (C. Hammond in The Basics of Crystallography and Diffraction, IUCr, Oxford Science Publications, Oxford, 2nd edn, 2001; International Tables for Crystallography Vol. A, ed. T. Hahn, The International Union of Crystallography, Kluwer Academic, Dordrecht, 4th edn, 1995. Aggregation into the highest symmetry is generally admitted for liquid crystalline materials. The symmetry of the observed liquid-crystalline cubic phase can thus characterized by a body-centered cubic network with the Im3m space group (no. 229) and a lattice parameter a = 47.3 Å (a = ∑hkl(h2 + k2 + l2)0.5 × dhkl/Nhkl]). As deduced from the calculated molecular volume (4660 Å3), the unit cell of the cubic lattice contains ≈20–24 C8amideNi mesogens.
- A. Escande, L. Guénée, E. Terazzi, T. B. Jensen, H. Nozary and C. Piguet, Eur. J. Inorg. Chem., 2010, 2746–2759 CrossRef CAS.
- F. Camerel, L. Bonardi, G. Ulrich, L. Charbonnière, B. Donnio, C. Bourgogne, D. Guillon, P. Retailleau and R. Ziessel, Chem. Mater., 2006, 18, 5009–5021 CrossRef CAS.
- M. J. Hostetler, J. J. Stokes and R. W. Murray, Langmuir, 1996, 12, 3604–3612 CrossRef CAS.
- S.-H. Park and C. E. Lee, Chem. Mater., 2006, 18, 981–987 CrossRef CAS.
- (a) D. J. P. Yeardley, G. Ungar, V. Percec, M. N. Holerca and G. Johansson, J. Am. Chem. Soc., 2000, 122, 1684–1689 CrossRef CAS; (b) H. Duan, S. D. Hudson, G. Ungar, M. N. Holerca and V. Percec, Chem.–Eur. J., 2001, 7, 4134–4141 CrossRef CAS; (c) I. Bury, B. Heinrich, C. Bourgogne, D. Guillon and B. Donnio, Chem.–Eur. J., 2006, 12, 8396–8413 CrossRef CAS; (d) X. Zeng, G. Ungar and M. Impéror-Clerc, Nat. Mater., 2005, 4, 562–567 CrossRef; (e) E. Terazzi, S. Torelli, G. Bernardinelli, J.-P. Rivera, J.-M. Bénech, C. Bourgogne, B. Donnio, D. Guillon, D. Imbert, J.-C. G. Bünzli, A. Pinto, D. Jeannerat and C. Piguet, J. Am. Chem. Soc., 2005, 127, 888–903 CrossRef CAS; (f) J.-K. Kim, M.-K. Hong, J.-H. Ahn and M. Lee, Angew. Chem., Int. Ed., 2005, 44, 328–332 CrossRef CAS; (g) T. Cardinaels, J. Ramaekers, P. Nockemann, K. Driesen, K. Van Hecke, L. Van Meervelt, G. Wang, S. De Feyter, E. Fernandez Iglesias, D. Guillon, B. Donnio, K. Binnemans and D. W. Bruce, Soft Matter, 2008, 4, 2172–2185 RSC; (h) S. Coco, C. Cordovilla, B. Donnio, P. Espinet, M. J. Garcia-Casas and D. Guillon, Chem.–Eur. J., 2008, 14, 3544–3552 CrossRef CAS.
- (a) A. M. Levelut and M. Clerc, Liq. Cryst., 1998, 24, 105–115 CrossRef CAS; (b) X. Zeng, G. Ungar and M. Impéror-Clerc, Nat. Mater., 2005, 4, 562–567 CrossRef.
- (a) S. Diele, Curr. Opin. Solid State Mater. Sci., 2002, 6, 333–342 Search PubMed; (b) S. Kutsumizu, Curr. Opin. Solid State Mater. Sci., 2002, 6, 537–543 CrossRef CAS; (c) M. Impéror-Clerc, Curr. Opin. Chem. Biol., 2005, 9, 370–376 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic details and characterizations of the benzil compounds and their corresponding metal complexes as well as DSC traces and XRD patterns of all the compounds, not presented in the main text. See DOI: 10.1039/c2ra20332d |
| This journal is © The Royal Society of Chemistry 2012 |