Green bromine: in situ generated catalyst for the selective oxidation of alcohols using H2O2 as a benign oxidant
Girdhar
Joshi
,
Rajendra D.
Patil
and
Subbarayappa
Adimurthy
*
Analytical Science Division, Central Salt & Marine Chemicals Research Institute, (Council of Scientific and Industrial Research), G.B. Marg, Bhavnagar, 364021, India. E-mail: sadimurthy@yahoo.com; Fax: (91)-278-2567562
First published on 17th January 2012
Abstract
The selective oxidation of benzylic/secondary alcohols to the corresponding aldehydes/ketones with a catalytic amount of bromide–bromate (10 mol%) couple and H2O2 as a benign oxidant has been developed. The oxidation reactions were achieved through acid (15 mol%) activation of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratios of a bromide–bromate couple for the in situ generation of the Br2/BrOH species. The In situ generation of the Br2/BrOH species is also supported by UV-Visible spectrophotometric studies. High selectivity and yields of aldehydes/ketones were obtained without the use of any transition metal catalyst under aqueous conditions.
:1 mole ratios of a bromide–bromate couple for the in situ generation of the Br2/BrOH species. The In situ generation of the Br2/BrOH species is also supported by UV-Visible spectrophotometric studies. High selectivity and yields of aldehydes/ketones were obtained without the use of any transition metal catalyst under aqueous conditions.
The oxidation of alcohols to carbonyl compounds is an important functional group transformation in synthetic chemistry. The conventional oxidation of alcohols predominantly involves transition metal catalysts which work as oxygen carriers. However, most of these catalysts require specific conditions like high temperatures, an inert reaction atmosphere, and occasionally co-catalysts. Furthermore the disposal of the unrecovered catalyst is also a matter of concern with respect to both the environment and the cost. Therefore the development of alternate reagents/catalysts to precious metal catalysts which can mediate the selective oxidation of alcohols is desirable. The halogen species (Cl, Br, and I) are frequently used for the oxidation of alcohols.1 Although, bromine is hazardous to handle, ship and use, it is still being used in industry as well as in academia due to its ready availability. Numerous methods have been reported for the oxidation of alcohols with varying amounts (2–40 mol%) of bromine or bromine based catalysts.2 Further, in some reported procedures, undesired oxidation products were observed.2
Conventional reagents employed for the oxidation of alcohols include permanganates,3 dichromates,4cerium salts,5nitric oxide,6 and other transition metal catalysts.7 During the last decade, a large number of metal catalysed oxidations have been disclosed and most of these oxidations were performed with complexes of precious metals.8 On the other hand, molecular O2 and H2O2 are cleaner oxidizing reagents.9,10 However, these reagents are not amenable to oxidise all the functional groups selectively. The hazardous effects associated with elemental bromine, the limited availability and high price of precious metals necessitate the continuous search for more economical and environmental friendly reagents/catalysts for such transformations. One such cost-effective oxidizing reagent is chlorine-gas/hypochlorite but its direct use in organic synthesis is limited due to the possibility of non-selective oxidation. Besides its direct use, chlorine/hypochlorite plays an important role in the preparation of other oxidizing agents.11 Additionally, there are useful halogen or halide derivatives such as oxy-halo compounds of bromine and iodine,12 and N-halosuccinimides13 are reported for the oxidation of alcohols. By choosing the right oxidizing reagent it is possible to control the oxidizing power and, consequently, the selectivity.
Assessment of results for halogen based oxidation of alcohols to aldehydes is presented in Table 1. As can be seen from the data of Table 1, most of the reported methods require stoichiometric or more than stoichiometric amounts of halogen containing reagents for oxidations along with other catalysts. In continuation of our efforts towards green and sustainable processes,14 we report in the present manuscript a selective oxidation of benzylic/secondary alcohols to corresponding carbonyl compounds using a bromide–bromate couple (10 mol%) with reference to alcohol under aqueous medium at room temperature (Scheme 1).
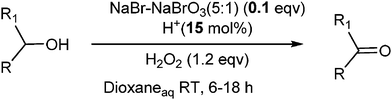
| Entry | Reagents | Reagent ratio (eqv.)a | Isolated yield (%) | Ref. |
|---|---|---|---|---|
| a Reagents used w.r.t. one eqv. of benzyl alcohol. b Yield based on GC conversion. IER = Ion exchange resin, MPHT = N-methylpyrrollidine-2-one hydro-tribromide, TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl, DBDMH= 1, 3-dibromo-5, 5 dimethyl hydantoin. | ||||
| 1 | NaBrO3/IER | 6.6 | 95 | 16 |
| 2 | Br2/PhIO2/NaNO2 | 0.02/0.01/0.01 | 94 | 17 |
| 3 | HBr/NaNO2 | 0.10/0.02 | 90 | 2n |
| 4 | NBS | 1 | 53.7b | 14g |
| 5 | NaIO4/H+ | 1/1 | 79 | 18 |
| 6 | N2H4·H2/I2 | 1/2 | 3.7 | 19 |
| 7 | KIO4/Et4NBr | 1/0.25 | 98 | 20 |
| 8 | MPHT/H2O2 | 0.1/2 | 93 | 21 |
| 9 | Br2/TEMPO/NaNO2 | 0.4/0.1/0.4 | 95 | 22 |
| 10 | DBDMH/TEMPO/NaNO2 | 40/0.1/0.4 | 88 | 23 |
| 11 | NaBr-NaBrO3/H+ | 0.05–0.4/0.15 | 84 | 14c |
| 12 | HBr/H2O2 | 0.1/1.2 | 78 | 14d |
| 13 | NaBr-NaBrO3/H+/H2O2 | 0.084–0.016/0.15/1.2 | 90 | Present |
We initiated the oxidation of benzylic/secondary alcohols using the in situ generation of catalytic Br2 (10 mol%) as a bromine source and H2O2 as a benign oxidant (eqn (1)–(3)). In this study, the bromide–bromate and acid generates brominein situ (eqn (1)), under aqueous conditions, bromine disproportionates to HBr and BrOH (eqn (2)).2m,15BrOH is revealed to be a mild and selective oxidizing agent for a number of organic substrates.
| 5Br− + BrO3− + 6H+→3Br2 + 3H2O | (1) |
| Br2 + H2O→BrOH + HBr | (2) |
| 2HBr + H2O2→Br2 + 2H2O | (3) |
In order to prove the formation of the BrOH species from the present system, we recorded the UV-absorption spectra of a mixture of bromide–bromate acid under aqueous medium in the absence (Fig. 1A) and presence (Fig. 1B) of an organic substrate according to the composition of equations 1–3. The absorption spectra were recorded at different concentrations (250, 300, 350 and 500 ppm) of bromide. As evidenced from the plot of the absorbance (Fig. 1A), it clearly shows the absorption bands at 220 nm, 266 nm and 390 nm correspond to HBr, BrOH and Br2 respectively.24,25 When the same spectra was recorded in the presence of an organic substrate (Fig. 1B) the same peaks at 220 nm and 266 nm correspond to HBr and BrOH were observed respectively. Initially the BrOH is observed clearly, as oxidation proceeds, HBr will be generated simultaneously with decrease of BrOH (Fig. 1B). In presence of H2O2, HBr oxidises to BrOH to continue the catalytic oxidation cycle as shown in Scheme 2.
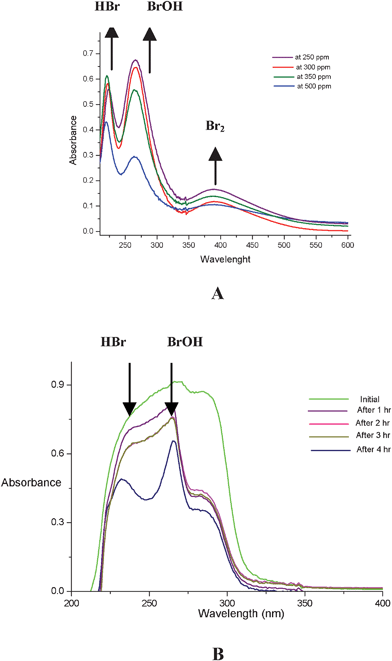 | ||
| Fig. 1 UV-Absorption spectra of BrOH (A) in absence and (B) in presence of organic substrate. | ||
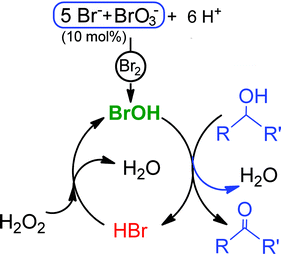 | ||
| Scheme 2 Proposed mechanism for the oxidation of alcohols. | ||
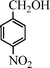
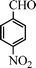
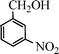
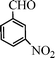
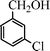
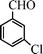
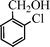
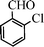
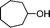
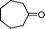
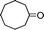
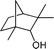
Under the present reagent conditions (Table 1, entry 13), benzaldehyde was isolated in 90% yield (Table 2, entry 1). p-Methyl benzyl alcohol provided a 94% yield with 100% selectivity (Table 2, entry 2). Benzylic alcohols with electron withdrawing nitro groups at positions (p/m/o) were selectively oxidized to the corresponding aldehyde in 95%, 93% and 75% isolated yields respectively (Table 2, entries 3–5). Similarly halogen (Br and Cl) substituted benzylic alcohols at different positions produced the corresponding aldehydes in good to excellent yields (88–95%) (Table 2, entries 6–9). The present protocol was extended towards the oxidation of secondary alcohols to the corresponding carbonyl compounds and the results are compiled in Table 3. As can be seen from the data of Table 3, various secondary alcohols such as cyclic, aryl and aryl-alkyl proceeded smoothly to provide the corresponding ketones in good yields (80–95%). To confirm the role of bromate, we carried out a controlled experiment without bromate on the oxidation of benzyl alcohol under similar reaction conditions to Table 1 with only 10 mol% of sodium bromide. We observed only 65% benzaldehyde formation during a period of 24 h against a 90% yield (Table 2, entry 1) in the presence of 1.6 mol% bromate. This study indicates that not only is a catalytic amount of bromate required for the efficient oxidation of alcohols, but it also significantly reduces the reaction time.
|
a Conditions: 1 eqv. alcohol, 5:1 NaBr–NaBrO3 (10 mol%), H+ (15 mol%) and 1.2 eqv. H2O2 in aq. Dioxane at r.t.
b Isolated yields after column chromatography.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Aldehydeb[Ref] |
| 1 |
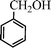
|
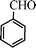
|
14 | 9014c,d |
| 2 |
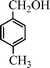
|
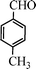
|
15 | 9414c,d |
| 3 |
|
|
18 | 9514c,d |
| 4 |
|
|
18 | 9314c,d |
| 5 |
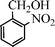
|
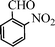
|
18 | 7514c,d |
| 6 |
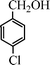
|
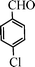
|
16 | 9514c,d |
| 7 |
|
|
16 | 9214c,d |
| 8 |
|
|
16 | 8814c,d |
| 9 |
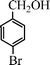
|
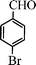
|
15 | 9514c,d |

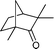
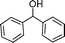
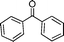
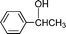
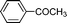
After the oxidation of alcohols to their corresponding carbonyl compounds, the bromide atoms end up in the aqueous effluent as HBr.26 The aqueous effluent containing HBr could be treated with H2O2 (eqn (3)) to regenerate the species (Br2/BrOH) for the effective oxidation of alcohols. In this manner the catalytic bromine generated in situ from the bromide–bromate couple accelerates the selective oxidation of alcohols to carbonyl compounds and makes the process environmentally friendly by dispensing in the use of precious/heavy metals.
To prove the recyclability of the reagent, an oxidation reaction was carried out at a 5.0 g (46.3 mmol) scale with benzyl alcohol as a substrate with 10 mol% of total bromide (5:1 NaBr:NaBrO3), 15 mol% acid (H2SO4) and 1.2 eqv. of 30% aqueous H2O2 under similar reaction conditions for a period of 16 h. The effluent obtained after the extraction of organic part was reused as the bromide source, to which a further calculated amount of substrate (5 g) and H2O2 (1.2 eqv. 30%) were added and the reaction was stirred for another 16 h. The same procedure was followed for three such consecutive runs and their conversion and turnover number (TON) are summarized in Table 4.
| Cycles | Yield (%)b | TON |
|---|---|---|
| a Reactions were carried out at a 5 g (46.3 mmol) scale of benzyl alcohol under similar reaction conditions. b Based on GC area%. [TON = (mmol. substrate/mmol. cat) conv.] | ||
| fresh | 88 | 880 |
| recycle 1 | 83 | 830 |
| recycle 2 | 80 | 800 |
| recycle 3 | 76 | 760 |
| recycle 4 | 73 | 730 |
In conclusion, we have described an efficient and selective method for the oxidation of benzylic/secondary alcohols to corresponding carbonyl compounds with catalytic bromine generated in situ as a halogen source and H2O2 as a cleaner and terminal oxidant under aqueous medium at room temperature without any transition metal catalyst. The bromide ion which ends up in the aqueous effluent is recyclable and can reused for the same oxidation process.
Acknowledgements
We thank Mr. Hitesh, Mr. V. Agrawal, Mr. H. Brahmbhatt and Mr. A.K. Das of this institute for supporting NMR, IR, GC-MS and LC-MS analyses respectively. G. J. and R. D. are thankful to CSIR, New Delhi, India for the award of SRF.References
- J. Palon, Chem. Soc. Rev., 1994, 23, 357 RSC
.
-
(a) F. Minisci, O. Porta, F. Recupero, C. Punta, C. Gambarotti, M. Pierini and L. Galimberti, Synlett, 2004, 2203 CrossRef CAS
-
(a) H. Firouzabadi, M. Maderi, A. Sardarian and M. Vessal, Synth. Commun., 1983, 13, 611 CrossRef CAS
- M. Z. Kassaee, S. Z. Sayyed-alangi and H. Sajjadi-Ghottbabadi, Molecules, 2004, 9, 825 CrossRef CAS
- D. N. Dhar and A. K. Bag, Indian J. Chem., 1984, 23B, 974 CAS
- W. A. Pryor, D. F. Church, C. K. Govindan and G. Crank, J. Org. Chem., 1982, 47, 156 CrossRef CAS
-
(a) J. C. Collins, W. W. Hess and F. J. Franck, Tetrahedron Lett., 1968, 9, 3363 CrossRef
-
(a) For palladium catalysts see: H. Wang, S. X. Deng, Z. R. Shen, J. G. Wang, D. T. Ding and T. H. Chen, Green Chem., 2009, 11, 1499 RSC
- V. Kesavan, D. Bonnet-Delpon and J. P. Begue, Synthesis, 2000, 223 CrossRef CAS
-
(a) J. Sebastian, K. M. Jinka and R. V. Jasra, J. Catal., 2006, 244, 208 CrossRef CAS
-
(a)
G. Ramachandraiah, P. K. Ghosh, S. Adimurthy, A. S. Mehta, A. D. Jethva and S. S. Vaghela, US Patent No. 6,740,253 dated 25 May, 2004 Search PubMed
-
(a) M. H. Ali and G. Bohnert, Synthesis, 1998, 1238 CrossRef
- N. S. Krishnaveni, K. Surendra and K. RamaRao, Adv. Synth. Catal., 2004, 346, 346 CrossRef CAS
-
(a) R. D. Patil, G. Joshi, S. Adimurthy and B. C. Ranu, Tetrahedron Lett., 2009, 50, 2529 CrossRef CAS
-
(a) A. Podgorsek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424 CrossRef CAS
- A. Shaabani and D. G. Lee, Synth. Commun., 2003, 33, 1255 CrossRef CAS
- R. Mu, Z. Liu, Z. Yang, Z. Liu, L. Wu and Z.-Li Liu, Adv. Synth. Catal., 2005, 347, 1333 CrossRef CAS
- T. M. A. Shaikh, L. Emmanuvel and A. Sudalai, J. Org. Chem., 2006, 71, 5043 CrossRef CAS
- P. Gogoi, G. K. Sarmah and D. Konwar, J. Org. Chem., 2004, 69, 5253 CrossRef
- A. R. Hajipour, F. Rafiee and A. E. Ruoho, Synth. Commun., 2006, 36, 2563 CrossRef CAS
- J. K. Joseph, S. L. Jain and B. Sain, Eur. J. Org. Chem., 2006, 590 CrossRef CAS
- D. Lenoir, Angew. Chem., Int. Ed., 2006, 45, 3206 CrossRef CAS
- R. Liu, C. Dong, X. Liang, X. Wang and X. Hu, J. Org. Chem., 2005, 70, 729 CrossRef CAS
-
Preparation of
BrOH
stock solution (250 ppm) for
UV-Vis
spectral study: A mixture of 27.0 mg (0.25 mmol) of NaBr and 8.0 mg (0.05 mmol) of NaBrO3 (5:1 NaBr/NaBrO3) were taken in a 100 mL standard volumetric flask and dissolved it with 2 mL water. To the above mixture, 23 mg (0.46 mmol) of H2SO4 and an aqueous solution of 30% H2O2 (3.75 mmol) was added, and the volume of flask was made up to the mark with water to obtain a concentration of 250 ppm. A similar procedure was followed to study the UV-Vis spectra in the presence of organic substrates (Fig. 1B).
-
(a) J. J. Orlando and J. B. Burkholder, J. Phys. Chem., 1995, 99, 1143 CrossRef CAS
- General Procedure for the oxidation of benzylic/ secondary alcohols ( benzyl alcohol as a representative substrate, Table 2 Entry 1): To a solution of benzyl alcohol (0.5 g, 4.625 mmol) in dioxane (2 mL) was added sodium bromide (0.040 g, 0.39 mmol) and sodium bromate (0.0117 g, 0.07 mmol) in water (1 mL). To the above mixture was added 15 mol% (0.04 g, 0.693 mmol) conc. H2SO4 (in 1 mL water) under stirring at room temperature during a period of 20 min followed by the slow addition of 1.2 eqv. of aqueous 30% H2O2 over a period of 2 h. The reaction mixture was stirred for a period of 12 h. The reaction was monitored by TLC. After the completion of the reaction, the product was extracted with ethyl acetate (5 mLx3). The combined organic extracts were washed with 5% sodium thiosulphate solution, and dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) and 90% (0.442 g) of benzaldehyde was obtained. The spectral data (NMR, IR, Mass etc.) were well matched with the literature data.14c,dSimilarly spectral data (NMR, IR, Mass etc.) of other products reported in Tables 2 and 3 were well matched with the reported data.
| This journal is © The Royal Society of Chemistry 2012 |