Investigation of the catalytic activity of poly(4-vinylpyridine) supported iodine as a new, efficient and recoverable catalyst for regioselective ring opening of epoxides
Nader Ghaffari
Khalig
*
Chemistry Department, University of Guilan, Rasht, PO Box 41335-19141, Iran. E-mail: ngkhaligh@gmil.com; ngkhaligh@guilan.ac.ir; Fax: +98 21 66934046; Tel: +98 21 66431738
First published on 23rd February 2012
Abstract
An efficient methodology for ring opening of epoxides in alcohols, water and acetic acid promoted by poly(4-vinylpyridine) supported iodine under neutral conditions, has been demonstrated. The reactions occur with high regioselectivity. The products were obtained in good to excellent yields. The reaction of optically active styrene oxide with P(4-VPI)+I3− in methanol was found to be very stereospecific and the product was isolated in 88% ee. This catalyst is stable and can be easily regenerated and used.
Introduction
Functional polymers are macromolecules to which, chemically, functional groups are attached; they have the potential advantages of small molecules with the same functional groups.1 Probably the most important advantage in using a functionalized polymer as a reagent or a catalyst is the simplification of product work-up, separation, and isolation. Supported reagents may also be used more conveniently in excess to drive reactions to completion, without incurring a penalty in work-up procedures. In addition, in most cases the used polymeric reagents can be regenerated. It is generally known that the efficiency of active functional groups attached to a polymeric support, either covalently or ionically, is affected by the structural features of the polymeric support such as polarity, swellability, nature and extent of cross-linking, etc.1–4Iodine has been attracting much attention since its discovery in 1811. It is the weakest oxidizer among the halogens and a poor electrophile that often needs the assistance of a strong acid or oxidizer. Iodine has several advantages over the vast majority of the other Lewis-acid catalysts, especially the metallic catalysts. Its catalytic potential is intriguingly broad; it is a water-tolerant, relatively cheap catalyst. Another distinctive feature of iodine is its high catalytic activity in dilute solutions, under highly concentrated reaction conditions (HCRC) as well as under solvent-free reaction conditions (SFRC). The latter reaction conditions are particularly important in terms of green chemistry; they contribute to waste-and health-hazard minimization and cost efficiency. For a long time iodine has been recognized as a good catalyst and reagent in carbohydrate chemistry.5,6 Different aspects of iodine chemistry have been reviewed; four reviews in Chinese7–10 on iodine as a catalyst11 and the use of iodine in protection–deprotection12 chemistry have been published.
Epoxides are versatile intermediates in synthetic organic chemistry and can be opened under a variety of conditions.13 The most practical and widely employed strategy for the synthesis of 1,2-bifunctional compounds was via nucleophilic ring opening using a Lewis acid or a strong base.14–20 Epoxide ring opening reactions to give β-substituted alcohols with carbon- and heteroatomic nucleophiles, rearrangement reactions providing carbonyl compounds, and isomerization reactions leading to allylic alcohols were useful tools in organic synthesis. Several reagents have been reported in the literature that can promote ring opening of epoxides.21–33 In most of the epoxide ring opening reactions under acidic conditions, the formation of a mixture of regioisomers and polymerization were observed. Some of the reported catalysts suffered from disadvantages such as high acidity, the non-catalytic nature of the reagents, long reaction times and inconvenient handling.34,35 Therefore, the introduction of new methods for the nucleophilic ring opening of epoxides, which work under mild conditions, is important in synthetic organic chemistry.
In continuation of our ongoing research program on the development of new catalyst and methods for organic transformations,36 in this study we decided to prepare quaternized poly(4-vinylpyridine) supported iodine and show that it can act as an efficient catalyst for ring opening of epoxides in alcohols, water and acetic acid. This material will be referred here as P(4-VPI)+I3− (Scheme 1). In the presence of the P(4-VPI)+I3−, the products were obtained with high regioselectivity under neutral conditions in high yields with the advantage of overcoming the problem of handling and sublimation of iodine.
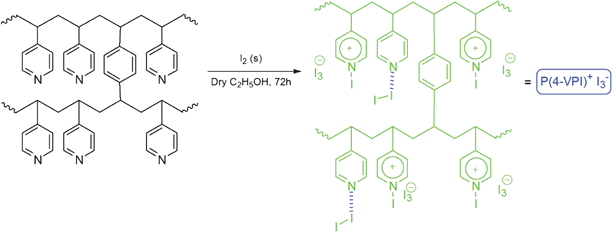 | ||
| Scheme 1 The preparation of the poly(4-vinylpyridine) supported iodine. | ||
Results and discussion
Studies to confirm the structure of the poly(N-iodo-4-vinylpyridinium)triodide
Fig. 1 presents the FTIR spectra of P(4-VP) and P(4-VPI)+I3−. In the spectrum of P(4-VP) the absorption bands at 1598.88, 1554.52, 1496.66, and 1413.72 cm−1 corresponding to νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and νCN stretching in pyridine ring appeared.37 On the other hand, the presence of an extra iodine atom on nitrogen atom of pyridine moiety in the P(4-VPI)+ increased the number of vibrational modes and brought completely different FTIR spectrum.38 The observed spectrum displayed the expected transitions due to P(4-VP), but also a second, prominent series of sharp transitions was observed, red-or blue-shifted similar to those of the P(4-VP) polymer (e.g. those related to the P(4-VP) at 997.13, 1068.49, and 1218.93 cm−1, see Fig. 1), but generally with larger absolute shifts. On the basis of a comparison with literature spectra,39 these transitions can be attributed to the poly(N-iodo-4-vinylpyridinium) cation P(4-VPI)+. As shown in Fig. 1 the bands at 1554.52 and 1413.72 cm−1 disappeared whereas a strong band at 1639.38 cm−1, related to P(4-VPI)+ appeared. The observed effect may be therefore ascribed to “some changes in aromatic ring” as a consequence of P(4-VPI)+ formation. Thus, treatment of P(4-VP) with ethanolic solution of iodine resulted in iodination of the pyridine units of the polymer.
C and νCN stretching in pyridine ring appeared.37 On the other hand, the presence of an extra iodine atom on nitrogen atom of pyridine moiety in the P(4-VPI)+ increased the number of vibrational modes and brought completely different FTIR spectrum.38 The observed spectrum displayed the expected transitions due to P(4-VP), but also a second, prominent series of sharp transitions was observed, red-or blue-shifted similar to those of the P(4-VP) polymer (e.g. those related to the P(4-VP) at 997.13, 1068.49, and 1218.93 cm−1, see Fig. 1), but generally with larger absolute shifts. On the basis of a comparison with literature spectra,39 these transitions can be attributed to the poly(N-iodo-4-vinylpyridinium) cation P(4-VPI)+. As shown in Fig. 1 the bands at 1554.52 and 1413.72 cm−1 disappeared whereas a strong band at 1639.38 cm−1, related to P(4-VPI)+ appeared. The observed effect may be therefore ascribed to “some changes in aromatic ring” as a consequence of P(4-VPI)+ formation. Thus, treatment of P(4-VP) with ethanolic solution of iodine resulted in iodination of the pyridine units of the polymer.
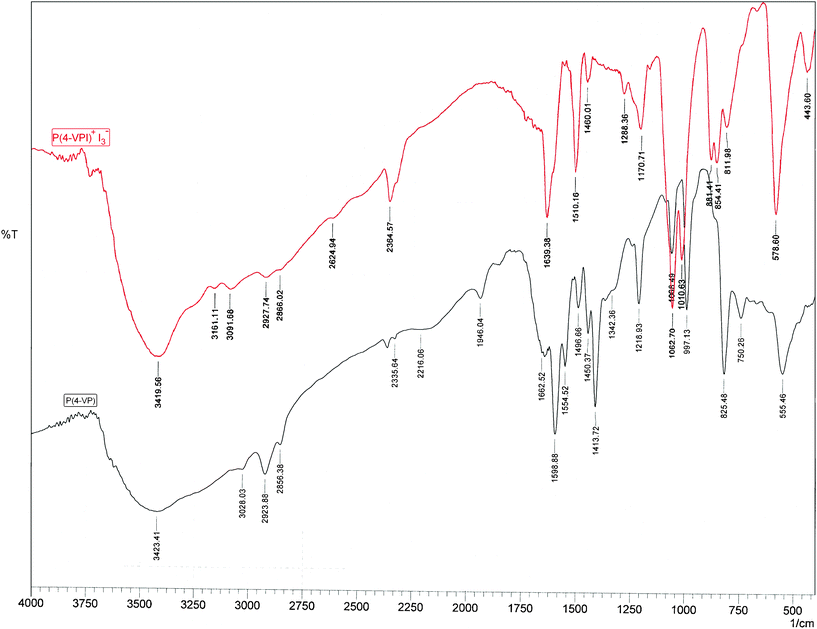 | ||
| Fig. 1 FTIR spectra of poly(4-vinylpyridine) (bottom) and poly(N-iodo-4-vinylpyridinium)triiodide (top). | ||
Reid and Mullikan40 reported in 1954 that the absorbance of the pyridine–iodine complex in pure pyridine (k = 290) in the 422 nm band shift slowly, increasing in intensity. They attributed it to the conversion of the initially-formed charge-transfer complex (outer complex) into an “inner complex”. Also ionic side reactions, such as
| (pyridine) + I |
| 2 |
| → [(pyridine)I] |
| + |
| + I |
| − |
| , |
| or 2(pyridine) + I2 → [(pyridine)2I]+ + I3−, |
The ring opening of epoxides in alcohols, water and acetic acid
At first, we examined the methanolysis of styrene oxide using various amounts of catalyst at room temperature. Although the reaction without a catalyst gave no product, it proceeded catalytically upon addition of catalyst. It showed that 10 mg (3.6 mol%) of P(4-VPI)+I3− was an adequate amount of the catalyst to conduct the ring opening reaction quantitatively to give 2-methoxy-2-phenyl ethanol as the sole product in 92% yield. In comparison, the ring opening behavior of styrene oxide with molecular iodine in the absence of the polymer, P(4-VP), in the absence of iodine and monomer pyridine–iodine solution is given in Table 1, entries 1, 2 and 3. As shown in Table 1 yield of methanolysis with this new supported catalyst was quite excellent (Table 1, entry 4).In order to show the general applicability of the method for the ring opening of epoxides with structurally different alcohols, water and acetic acid, we studied the reaction of the solvolysis of different classes of epoxides. We applied similar reaction conditions for the solvolysis of the epoxides in the presence of this catalyst. NMR and GC analysis showed that the reactions were highly regioselective in the presence of P(4-VPI)+I3−.
In order to show the effect of increased temperature, the reaction of methanolysis of 1,2-epoxy-3-phenoxypropane (1b) and 1,2-epoxycyclohexane (1c) were studied (Table 2, entries 8, 9 and 16, 17). As shown, the reaction rates were considerably increased but the reaction yields were slightly increased.
| Entry | Epoxide | Solvent | Product (ratio 2g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2h)a :2h)a |
Time (min)/T (°C) | Yield (%)b |
|---|---|---|---|---|---|
| a All products were identified by comparison of their physical and spectral data with those of authentic samples.37,39 b GC yields. c R(+)-styrene oxide [α]D = +33.0 (undiluted), from Aldrich, was used without further purification. d Value not corrected for the optical purity of the starting material and determined according to the literature.42 | |||||
| 1 | 1a | MeOH | 100:0 |
20 (20, 25, 25)/25 | 92 (92,90,86) |
| 2 | 1a | EtOH | 100:0 |
20/25 | 80 |
| 3 | 1a | n-PrOH | 100:0 |
20/25 | 81 |
| 4 | 1a | i-PrOH | 100:0 |
28/25 | 80 |
| 5 | 1a | t-BuOH | 100:0 |
32/25 | 75 |
| 6 | 1a | H2O–MeCN | 1,2-diol | 32/60 | 90 |
| 7 | 1a | AcOH | 100:0 |
16/60 | 68 |
| 8 | 1b | MeOH | 0:100 |
16/25 | 82 |
| 9 | 1b | MeOH | 0:100 |
10/65 | 94 |
| 10 | 1b | EtOH | 0:100 |
25/65 | 86 |
| 11 | 1b | n-PrOH | 0:100 |
22/65 | 85 |
| 12 | 1b | i-PrOH | 0:100 |
25/65 | 81 |
| 13 | 1b | t-BuOH | 0:100 |
28/65 | 67 |
| 14 | 1b | H2O–MeCN | 1,2-diol | 32/65 | 94 |
| 15 | 1b | AcOH | 0:100 |
32/65 | 86 |
| 16 | 1c | MeOH | trans | 25/25 | 82 |
| 17 | 1c | MeOH | trans | 15/65 | 87 |
| 18 | 1c | EtOH | trans | 15/65 | 82 |
| 19 | 1c | n-PrOH | trans | 28/65 | 80 |
| 20 | 1c | i-PrOH | trans | 32/65 | 78 |
| 21 | 1c | t-BuOH | trans | 32/65 | 58 |
| 22 | 1c | H2O–MeCN | trans | 32/65 | 89 |
| 23 | 1c | AcOH | trans | 26/65 | 89 |
| 24 | 1d | MeOH | 0:100 |
25/25 | 87 |
| 25 | 1d | EtOH | 0:100 |
28/65 | 80 |
| 26 | 1d | n-PrOH | 0:100 |
28/65 | 80 |
| 27 | 1d | i-PrOH | 0:100 |
28/65 | 78 |
| 28 | 1d | t-BuOH | 0:100 |
32/65 | 75 |
| 29 | 1d | H2O–MeCN | 1,2-diol | 32/65 | 76 |
| 30 | 1d | AcOH | 0:100 |
28/65 | 85 |
| 31 | 1e | MeOH | 0:100 |
23/25 | 84 |
| 32 | 1e | EtOH | 0:100 |
38/65 | 88 |
| 33 | 1e | n-PrOH | 0:100 |
38/65 | 82 |
| 34 | 1e | i-PrOH | 0:100 |
40/65 | 78 |
| 35 | 1e | t-BuOH | 0:100 |
45/65 | 70 |
| 36 | 1e | H2O–MeCN | 1,2-diol | 45/65 | 82 |
| 37 | 1e | AcOH | 0:100 |
32/65 | 82 |
| 38 | 1f | MeOH | 0:100 |
50/25 | 80 |
| 39 | 1f | EtOH | 0:100 |
50/65 | 84 |
| 40 | 1f | n-PrOH | 0:100 |
55/65 | 81 |
| 41 | 1f | i-PrOH | 0:100 |
55/65 | 78 |
| 42 | 1f | t-BuOH | 0:100 |
55/65 | 72 |
| 43 | 1f | H2O–MeCN | 1,2-diol | 70/65 | 82 |
| 44 | 1f | AcOH | 0:100 |
50/65 | 88 |
| 45 | R-(+)-styrene oxide | MeOH | S-(+)-2-methoxy-2-phenyl ethanolc | 65/25 | 92 (88% e.e.)d |
Also shown, the polarity of solvent has a slight effect on the rate and yield of the reactions (Table 2, entries 1,5). The proticity of the solvent decreased the rate of the reaction and slightly effected on yield of the reaction (Table 2, entries 1,7). The explanation for this result may be due to the protonation of pyridine moieties of polymer and also there can be a possibility of formation hydrogen iodide in the reaction as by-product which will reduce the reactivity.
In order to see the effect of polar electron-withdrawing groups adjacent to the epoxide's ring, the reactions of 1,2-epoxy-3-chloropropane (1f) were studied (Scheme 2). As expected, the reaction rates were considerably decreased. All of these reactions were performed at 65 °C (Table 2, entries 38–42). In all of these reactions only one regioisomer was obtained.
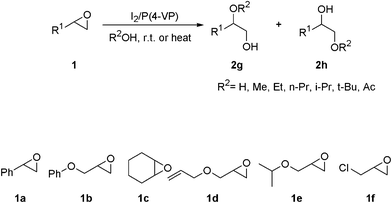 | ||
| Scheme 2 The ring opening the epoxides in different alcohols, water and acetic acid. | ||
For 1,2-epoxy-3-isopropoxypropane (1e), which has a deactivating group containing an ethereal linkage, all of the reactions were performed at 65 °C (Table 2, entries 32–37). Cleavage of ethereal linkage was not observed in these reactions.
Reactions of styrene oxide as an active epoxide were carried out at room temperature with methanol, ethanol, 1-propanol, 2-propanol and 2-methyl-2-propanol in the presence 10 mg (∼0.036 mmol) of P(4-VPI)+I3−, which afforded 2-alkoxy-2-phenyl ethanol in 75–94% isolated yields within 20–32 min with trace formation of other regioisomers (Table 2, entries 1–5).
The reactions of 1,2-epoxycyclohexane (Scheme 2, 1c) were slightly slower than those of styrene oxide. Although the reactions of primary and secondary alcohols were nearly complete in about 15–32 min in the presence of 10 mg of the catalyst at room temperature or 65 °C (Table 2, entries 16–21), in 2-methyl-2-propanol a decrease in the reaction rate was observed and trans-2-t-butoxycyclohexanol was obtained after 32 min in only 58% yield. In all of the reactions involving cyclohexene oxide only trans products were obtained.
Alcoholysis of other epoxides, such as 1,2-epoxy-3-phenoxypropane (1b) and 1,2-epoxy-3-allyloxypropane (1d) were performed (Table 2, entries 8–13 and 24–29). The results showed that the ethereal linkage and the double bond remain intact during the course of the alcoholysis reaction of the epoxide ring. For these unsymmetrical epoxides, the alkoxy group is incorporated preferentially at the less-hindered epoxide carbon atom.
Epoxides were converted to the corresponding diols as the sole product in the presence of 10 mg of P(4-VPI)+I3− in an acetonitrile–water mixture (Table 2, entries 6, 14, 22, 29, 36, 43). All of these reactions were performed at 65 °C.
Different classes of epoxides were converted to their corresponding β-acetoxy alcohols in the presence of 10 mg P(4-VPI)+I3− as a catalyst in acetic acid (Table 2, entries 7,15,23,30,37,44). The reactions of epoxides required high temperature (65 °C) for complete conversion to their corresponding β-acetoxy alcohols. As shown in Table 2, a highly regioselective formation of β-acetoxy alcohols was observed in all cases involving unsymmetrical epoxides.
In the case of unsymmetrical epoxides, the reactions were regioselective, and the attack of the nucleophile on the less substituted oxirane carbon yielded 2h type products (Scheme 2). The only exception was the alcoholysis and acetolysis of styrene oxide, in which the reactions occurred on the more substituted carbon and 2g type products were produced (Scheme 2). In the case of active epoxides and when the alcohol has a higher nucleophilicity, the reactions were carried out at room temperature.
As shown in Table 2, solvolysis of alkyl substituted epoxides occurs much faster than those epoxides carrying an electron-withdrawing substituent. Identification of these compounds was done by comparison with authentic samples, which were prepared according to known procedures.37,39
In order to elucidate the stereospecificity of the P(4-VPI)+I3− catalyzed ring opening of epoxides, the reaction of optically active R(+)-styrene oxide was studied with iodine (9 mg ∼0.036 mmol) and P(4-VPI)+I3− (10 mg, ∼0.036 mmol) (Table 2, entry 45). We observed that R-(+)-styrene oxide (> 98% e.e.) reacts enantioselectivity with inversion of configuration on the benzylic center to provide S-(+)-2-methoxy-2-phenyl ethanol (92%, 88% e.e.). The highest optical purity was obtained in an enantiopure form in high yield and under high regioselectivity by using P(4-VPI)+I3− at room temperature. The optical rotation of the product was determined and compared with that reported in the literature.42 Using elemental iodine for methanolysis of R-(+)-styrene oxide (> 98% e.e.) at room temperature, the optical purity of the product obtained was not high (34.4% e.e.).
The competitive ring opening reaction of styrene oxide (1a) and 1,2-epoxycyclohexane (1c) in the presence of the 1,2-epoxy-3-phenoxypropane (1b), 1,2-epoxy-3-allyloxypropane (1d), 1,2-epoxy-3-isopropoxypropane (1e) and 1,2-epoxy-3-chloropropane epoxides (1f), was observed with very high selectivity (Table 3, entries 1–8). Styrene oxide (1a) and 1,2-epoxycyclohexane (1c) reacted quantitatively while epoxides carrying electron-withdrawing substituents and deactivating groups (1b, 1d, 1e and 1f) remained intact. However, the reaction of styrene oxide (1a) in presence of 1,2-epoxycyclohexane (1c) in methanol show no selectivity under these conditions (Table 3, entry 9).
| Entry | Epoxide mixture | Time (min)/T (°C) | % Conversion of epoxidea | |
|---|---|---|---|---|
| a GC conversion (the percentage of epoxide that has reacted). | ||||
| 1 | 1a + 1b | 25/25 | 100 (1a) | 0.0 (1b) |
| 2 | 1a + 1d | 25/25 | 100 (1a) | 0.0 (1d) |
| 3 | 1a + 1f | 40/25 | 100 (1a) | 0.0 (1f) |
| 4 | 1a + 1e | 40/25 | 100 (1a) | 0.0 (1e) |
| 5 | 1c + 1b | 25/25 | 100 (1c) | 0.0 (1b) |
| 6 | 1c + 1d | 25/25 | 100 (1c) | 0.0 (1d) |
| 7 | 1c + 1f | 40/25 | 100 (1c) | 0.0 (1f) |
| 8 | 1c + 1e | 40/25 | 100 (1c) | 0.0 (1e) |
| 9 | 1a + 1c | 25/25 | 100 (1a) | 62 (1c) |
In order to show the merit of the presented protocol for the ring opening of epoxides with alcohols, we have compared the results obtained using P(4-VPI)+I3− with some of those reported in the literature for the reaction of 1,2-epoxy-3-phenoxypropane (1b) with methanol as tabulated in Table 4.
A plausible mechanism for the ring opening of epoxides
Although the precise mechanism of the reaction awaits further studies, a possible mechanistic pathway for the ring opening of epoxides, which rationalizes the formation of products, was presented in Scheme 3. On the basis of literature evidence,33,41,43–47 the first step involved the formation of a 1:1 molecular complex between P(4-VP) and elemental iodine (outer complex). This complex was further dissociated to release poly(N-iodo-4-vinylpyridinium) cation and triiodide anion (inner complex). Therefore, in this way, molecular iodine was converted to poly(N-iodo-4-vinylpyridinium) cation and polyiodide anion I2n+1− in the presence of the P(4-VP) and, in the second step, the poly(N-iodo-4-vinylpyridinium) and polyiodide ions participated in the ring opening of the epoxides. Polyiodides were capable of complexing oxygen functional groups. Ring opening of epoxides can occur via two mechanistic pathways, either nucleophilic attack by triiodide anion on the less sterically hindered epoxide carbon or via nucleophilic attack by O atom of hydroxyl group on P(4-VPI)+-epoxide complex, giving the transition state (A) (Scheme 3). On the other hand, electrophilic attack by molecular iodine, behaving as Lewis acid, leads to a mixture of 2g and 2h products. According to the mechanism, in P(4-VP) complexation with molecular iodine and, hence, elaboration of triiodide ion occurred much faster than the other way. After work-up, the catalyst could be recovered easily and could be reused several times.
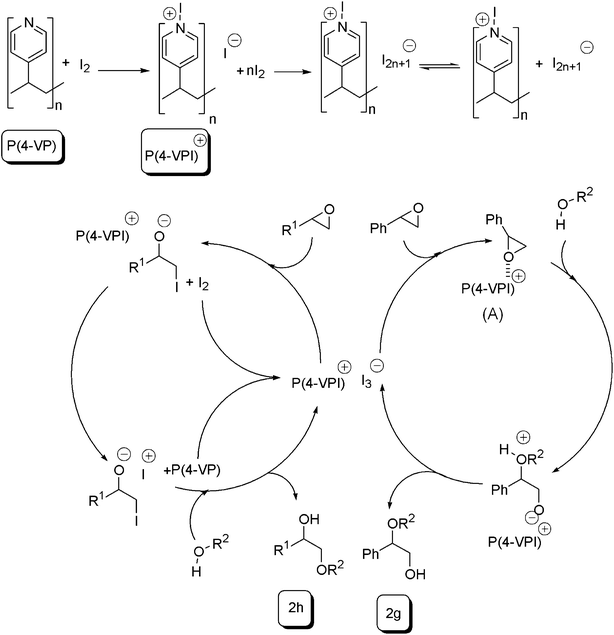 | ||
| Scheme 3 A plausible mechanism for the reaction. | ||
Recovering and reusing of the catalyst
To raise this method’s worth, the reusability of it was studied. For this purpose, the ring opening of epoxide 1a using P(4-VPI)+I3− was carried out several times, and the reaction mixtures were combined. Afterward, the solvent was evaporated, Et2O was added to the combined reaction mixtures, stirred for 10 min, and the catalyst was collected by filtration, washed with Et2O, dried at ambient temperature and used again in later reactions. The same procedures were repeated four times; however, as shown in Table 2 entry 1, even after four cycles, it still has excellent yields. It implied that the catalytic system can be reused at least four times without significant loss of activity. FTIR spectra of catalyst after fourth run show that interaction between I2 and the polymer was enough strong, the leaching of I2 could be connived. Moreover, the catalyst shows good stability in air versus time and temperature. It seem reasonable to expect that the structure stability of P(4-VPI)+I3− results from n-donor action at the N atom of pyridyl groups.Experimental
Substrates were purchased from Fluka AG and Merck Companies. Cross-linked poly(4-vinylpyridine) (2% divinylbenzene as cross-linking agent) was a commercial product (Fluka). All yields refer to isolated products. All products were known compounds, and they were identified by comparison of their spectra and physical data with those of the authentic samples. Melting points were determined in open capillaries on a Buchi 510 apparatus. IR spectra were recorded on a Perkin Elmer 781 spectrometer. NMR spectra were recorded on a Bruker Advance DPX 300 MHz spectrometer. Mass spectra were recorded on a Shimadzu GCMS-QP 1000 EX. Determination of the purity of the substrate and reaction monitoring were accomplished by TLC or by GLC on a Shimadzu model-GC-8A instrument with a flame ionization detector and using a column of 15% carbowax 20 M on chromosorb-w acid washed 60–80 mesh.Procedure for preparation of the poly(N-iodo-4-vinylpyridinium)triiodide
Cross-linked poly(4-vinylpyridine) (1.0 g) was suspended in ethanol (10 mL) and allowed to swell for about 24 h at room temperature. To this suspension, iodine (2.0 g, 7.87 mmol) was slowly added and the reaction mixture was heated at 50–55 °C for 48 h. An additional portion of iodine (1.0 g, 3.93 mmol) was added and the heating was continued for about 24 h. The reaction mixture was then filtered, washed with distilled water (3 × 10 mL) and diethyl ether (2 × 5 mL). The black violet solid, P(4-VPI)+I3− was dried in the presence of P4O10 under vacuum at 40 °C overnight to give the product, 2.6 g. The amount of iodide was determined by gravimetric and potentiometric titration methods,48 and was 3.62 mmol I− per gram of polymer.General procedure for the reaction of epoxides with alcohols, water and acetic acid in the presence P(4-VPI)+I3−
P(4-VPI)+I3− (10 mg, ∼3.6 mol%) was added to a solution of epoxide (1 mmol) in the methanol, water or acetic acid (2 ml). The reaction mixture was stirred for the specified time and the appropriate temperature according to Table 2. The progress of the reaction was monitoring by TLC. Solvent was evaporated, ether (10 mL) was added, and the catalyst was filtered. After extraction of the products, the catalyst was collected, dried to give 9.87 mg (98.7% recovered) and reused for the next run. The filtrate was washed with 10% aqueous solution of Na2S2O3 until disappearance of the iodine color was observed. The aqueous layer was separated and washed with ether (3 × 10 mL). Evaporation of the organic solution followed by column chromatography on a short column of silica gel afforded the pure product.Conclusion
In conclusion, P(4-VPI)+I3− can be regarded as neutral catalysts or reagents for efficient ring opening of epoxides. Due to this neutrality, they may find application for ring the opening of those epoxides which contain acid-sensitive functional groups. P(4-VP) supported iodine is very stable, nonvolatile, and its toxicity is greatly reduced. Availability, ease of procedure, reusability, high regioselectivity, chemoselectivity, mild reaction conditions, and high yields of products indicate a new synthetic application for heterogeneous catalytic iodine in organic synthesis. In comparison with iodine, the reactions of P(4-VPI)+I3− with optically active styrene oxide are more enantioselective. The most important advantages of this catalyst is that in all cases the product selectivity is 100%, while using other Lewis acids in the alcoholysis of epoxides, two products have been detected in the reaction mixture.Acknowledgements
I would like to acknowledge the support of this work by Guilan University research council.References
- A. Akelah and D. C. Sherindton, Application of functionalized polymers in organic synthesis,, Chem. Rev., 1981, 81, 557–587 CrossRef CAS.
- D. C. Sherington and P. Hodge, Synthesis and Separation Using Functional Polymers, Wiley, Chichester. 1988 Search PubMed.
- K. Takemoto, Y. Inaki and R. M. Otten brite, Functional Monomers and Polymers, Dekker, New York. 1987 Search PubMed.
- S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leac, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, Multi-step organic synthesis using solid-supported reagents and scavengers: A new paradigm in chemical library generation,, J. Chem. Soc., Perkin Trans. 1, 2000, 3815–4195 RSC.
- A. R. Vaino and W. A. Szarek, Iodine in carbohydrate chemistry., Adv. Carbohydr. Chem. Biochem., 2001, 56, 9–63 CrossRef CAS.
- A. Biswas, G. S. Selling, R. L. Shogren, J. L. Willett, C. M. Buchanan and H. N. Cheng, Iodine-catalyzed esterification of polysaccharides, Chim. Oggi Chem. Today., 2009, 27, 33–35 CAS.
- H.-S. Wang, J.-Y. Miao and L.-F. Zhao, Chin. J. Org. Chem., 2005, 25, 615–618 CAS.
- Z. Zhang and Q. Liu, Progress Chem., 2006, 18, 270–280 CAS.
- S. Shen, X. Xu and S. Ji, Chin. J. Org. Chem., 2009, 29, 806–811 CAS.
- Y. Zhou, P. Yan, G. Li and Z. Chen, Chin. J. Org. Chem., 2009, 29, 1719–1727 CAS.
- A. K. Banerjee, W. Vera, H. Mora, M. S. Laya, L. Bedoya and E. V. Cabrera, J. Sci. Ind. Res., 2006, 65, 299–308 CAS.
- S. Das, R. Borah, R. R. Devi and A. Thakur, Synlett, 2008, 2741–2762 CAS.
- J. G. Smith, Synthesis, 1984, 629–656 CrossRef CAS.
- H. Sharghi, N. Ali, M. Nasseri and K. Niknam, J. Org. Chem., 2001, 66, 7287–7293 CrossRef CAS.
- D. M. Hodgson, A. R. Gibbs and G. P. Lee, Tetrahedron, 1996, 52, 14361–14384 CrossRef CAS.
- I. Paterson and D. J. Berrisford, Angew. Chem., Int. Ed. Engl., 1992, 31, 1197–1202 CrossRef.
- S. K. Taylor, Tetrahedron, 2000, 56, 1149–1163 CrossRef CAS.
- R. E. Parker and N. S. Isaacs, Chem. Rev., 1959, 59, 737–799 CrossRef CAS.
- A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron, 1983, 39, 2323–2367 CrossRef CAS.
- N. Iranpoor and I. Mohammadpour-Baltork, Synth. Commun., 1990, 20, 2789–2797 CrossRef CAS.
- (a) N. Iranpoor, B. Tamami and K. Niknam, Can. J. Chem., 1997, 75, 1913–1919 CrossRef CAS; (b) H. Firouzabadi, N. Iranpoor, A. A. Jafari and S. Makarem, J. Mol. Catal. A: Chem., 2006, 250, 237–242 CrossRef CAS; (c) N. Iranpoor, M. Shekarriz and F. Shiriny, Synth. Commun., 1998, 28, 347–366 CrossRef CAS.
- V. Mirkani, S. Tangestaninejad, B. Yadollahi and L. Alipanah, Tetrahedron, 2003, 59, 8213–8218 CrossRef.
- B. M. Cheudary and Y. Sudha, Synth. Commun., 1996, 26, 2989–2992 CrossRef.
- N. Iranpoor and P. Salehi, Synthesis, 1994, 1152–1154 CrossRef CAS.
- R. V. Yarapathi, S. M. Reddy and S. Tammishetti, React. Funct. Polym., 2005, 64, 157–161 CrossRef CAS.
- J. Barluenga, H. Vazquez-Villa, A. Ballesteros and J. Gonzalez, Org. Lett., 2002, 4, 2817–2819 CrossRef CAS.
- K. Fagnou and M. Lautens, Org. Lett., 2000, 2, 2319–2321 CrossRef CAS.
- S. Tangestaninejad, M. Moghadam, V. Mirkhani, B. Yadollahi and S. M. R. Mirmohammadi, Monat. Fur Chem., 2006, 137, 235–242 CrossRef CAS.
- N. Iranpoor and H. Adibi, Bull. Chem. Soc. Jpn., 2000, 73, 675–680 CrossRef CAS.
- I. Mohammadpoor-Baltork, S. Tangestaninejad, H. Aliyan and V. Mirkhani, Synth. Commun., 2000, 30, 2365–2374 CrossRef CAS.
- M. Moghadam, I. Mohammadpoor-Baltork, S. Tangestaninejad, V. Mirkhani, L. Shariati, M. Babaghanbari and M. Zarea, J. Iran. Chem. Soc., 2009, 6, 789–799 Search PubMed.
- E. N. Jacobsen, F. Kakiuchi, R. G. Konsler, J. F. Larrow and M. Tokunaga, Tetrahedron Lett., 1997, 38, 773–776 CrossRef CAS.
- H. Sharghi, K. Niknam and M. Pooyan, Tetrahedron, 2001, 57, 6057–6064 CrossRef CAS.
- G. A. Olah, A. P. Fung and D. Mieder, Synthesis, 1981, 280–282 CrossRef CAS.
- G. H. Posner, D. Z. Rogers, C. H. Kinzig and G. M. Gurria, Tetrahedron Lett., 1975, 16, 3597–3600 CrossRef.
- (a) N. G. Khaligh and F. Shirini, J. Mol. Catal. A: Chem., 2011, 348, 20 CrossRef CAS; (b) N. G. Khaligh, J. Mol. Catal. A: Chem., 2011, 349, 63–70 CrossRef CAS; (c) F. Shirini and N. G. Khaligh, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2156–2165 CrossRef CAS; (d) N. G. Khaligh, Ultrason. Sonochem., 2011 DOI:10.1016/j.ultsonch.2011.12.004; (e) F. Shirini and N. G. Khaligh, Monatsh. Chem., 2011 DOI:10.1007/s00706-011-0612-5; (f) F. Shirini and N. G. Khaligh, J. Iran. Chem. Soc., 2012 DOI:10.1007/s13738-011-0060-5.
- B. Grzyb, J. Machnikowski and J. V. Weber, J. Anal. Appl. Pyrolysis, 2004, 72, 121–130 CrossRef CAS.
- L. N. Karklin, M. W. Kliujev and A. D. Pomogailo, Kin. Kat., 1983, 24, 408–412 Search PubMed.
- (a) B. Jones, G. J. Moody and J. D. R. Thomas, Inorg. Chem., 1970, 9, 114–119 CrossRef CAS; (b) I. Haque and J. L. Wood, Spectrochim. Acta., 1967, 23A, 959–967 CrossRef; (c) R. A. Zingaro and W. E. Tolberg, J. Am. Chem. Soc., 1959, 81, 1353–1357 CrossRef CAS.
- C. Reid and R. S. Mulliken, J. Am. Chem. Soc., 1954, 76, 3869–3874 CrossRef CAS.
- T. Tassaing and M. Besnard, J. Phys. Chem. A, 1997, 101, 2803–2808 CrossRef CAS.
- C. Jaramillo, J. L. Chiara and M. Martin-Lomas, J. Org. Chem., 1994, 59, 3135–3142 CrossRef CAS.
- L. F. Audrieth and E. J. Birr, J. Am. Chem. Soc., 1933, 55, 668–673 CrossRef CAS.
- G. Kortum and H. Wilskiz, Z. Physik. Chem., 1953, 202, 35–42 CAS.
- M. I. Konnaklieva, M. L. Dahi and E. Turos, Tetrahedron Lett., 1992, 33, 7093–7096 CrossRef.
- (a) H. Sharghi, A. R. Massah and M. Abedi, Talanta, 1999, 49, 531–538 CrossRef CAS; (b) A. Semnani and M. J. Shamsipur, J. Chem. Soc., Dalton Trans., 1996, 2215–2218 RSC; (c) M. R. Gangali, H. Sharghi, H. Eshghi and M. Shamsipur, J. Electroanal. Chem., 1996, 405, 177–181 CrossRef.
- H. Sharghi, M. Eskandari and R. Ghavami, J. Mol. Catal. A: Chem., 2004, 215, 55–62 CrossRef CAS.
- W. J. William, HandBook of Anion Determination, First ed. 1979, pp. 416–421 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |