Brønsted acid-promoted dimerization of o-alkynylbenzaldehydes: a one-step synthesis of functionalized Kagan's ether analogues†‡
Hao
Zhang
,
Wei-Chen
Cui
,
Zhi-Long
Hu
,
Shu-Yan
Yu
,
Shaozhong
Wang
* and
Zhu-Jun
Yao
*
State Key Laboratory of Coordination Chemistry and Nanjing National Laboratory of Microstructures, Institute of Chemical Biology and Drug Innovation, School of Chemistry and Chemical Engineering, Nanjing University, 22 Hankou Road, Nanjing, Jiangsu 210093, China. E-mail: wangsz@nju.edu.cn.; yaoz@nju.edu.cn
First published on 13th April 2012
Abstract
The Brønsted acid-promoted dimerization of o-alkynylbenzaldehydes has been discovered and studied in the presence of 45% aq. HBF4 in acetic acid. The developed cascade methodology provides a convenient one-step synthesis of symmetrical 2,3,6,7-dibenzo-9-oxabicyclo[3.3.1]nona-2,6-diene (Kagan's ether) analogues bearing various functionalities.
2,3,6,7-Dibenzo-9-oxabicyclo[3.3.1]nona-2,6-dienes and their derivatives (Kagan's ethers, Fig. 1)1,2 are a unique category of oxygenated heterocycles possessing anti-inflammatory and HIV-1 inhibitory activities.3,4 Because of their unique symmetric structures and interesting biological properties, the synthesis of Kagan's ethers has attracted considerable attention from the synthetic organic community since Kagan's pioneering work.2 A considerable number of useful methods have been developed for the synthesis of such oxa bridged-ring structures in recent decades.5–9
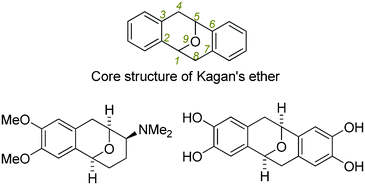 | ||
| Fig. 1 Core structure of Kagan's ethers, and two representative bioactive derivatives. | ||
During our recent investigation of isochromenylium tetrafluoroborates (ICTBs), a new type of air-stable oxonium salt having non-classical Hückel aromaticity, we found that the o-alkynylbenzaldehydes bearing stronger electron-donating group(s) in their A benzene ring could be easily converted into the corresponding ICTBs (represented by 2a in Fig. 2) by treatment with 45% aq. tetrafluoroboric acid in acetic acid under mild conditions.10,11 However, our further exploration of the substrate scope of these reactions showed that the simplest o-alkynylbenzaldehyde 1b (lacking a substituent in the benzene ring A) could not undergo this transformation at room temperature. When the temperature was raised to 75 °C, the reaction was promoted and completed in 20 h, affording an unusual white solid (2b) in 60% yield. Analysis of its 1H NMR, 13C NMR and mass spectra indicated that it was a symmetrical dimer derived from the o-alkynylbenzaldehyde 1b. The structure of 2b was determined by 2D NMR methods and finally confirmed by X-ray single crystallographic analysis (Fig. 2). Compound 2b features all the structural characters of Kagan's ether with two acyl substituents at the C4 and C8 positions. To examine the possible generality of temperature effects, the previously studied substrate having two dimethoxy substituents on the benzene ring A, o-alkynylbenzaldehyde 1a was also treated with 45% aq. tetrafluoroboric acid in acetic acid at 75 °C. Aldehyde 1a disappeared in 3 h and solid ICTB 2a was collected in 86% yield, which was similar to the reaction at room temperature. Furthermore, heating ICTB 2a with 45% aq. tetrafluoroboric acid in acetic acid at 75 °C for 20 h did not afford any dimeric product(s). Based on these primary findings, we speculated that the different electronic properties of the benzene ring A of substrates 1a and 1b could be important for determining the two discriminate reaction pathways, and that a new multistep cascade could be involved in the dimerization of 1b. Considering the biological importance of Kagan's ethers and their analogues, we further explored the reaction generality of this dimerization, since it would enable us to synthesize Kagan's ether in a single step. Herein, we report our results on the study of the dimerization of o-alkynylbenzaldehydes under Brønsted acid-promoted conditions.
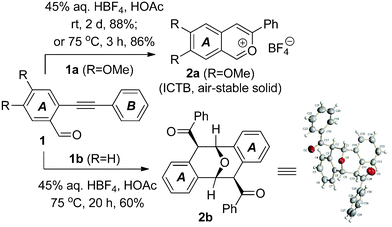 | ||
| Fig. 2 Transformation of o-alkynylbenzaldehyde 1b to Kagan's ether derivative 2b. | ||
Initial optimization of reaction conditions was conducted. Using the o-alkynylbenzaldehyde 1b as a substrate and 45% aq. tetrafluoroboric acid as the Brønsted acid, several commonly used protic acids were examined as acidic media (Table 1). For screenings of the stronger Brønsted acids TfOH and TFA, their dilute solutions in neutral solvents were employed as the acidic media. Lower yields of product 2b were obtained in both reactions in 1,2-dichloroethane at 75 °C in the presence of TFA (1 equiv.) and TfOH (1 equiv.), respectively (entries 10 and 14). Reactions in acetic acid were found to be cleaner and afforded much higher yields of 2b (entries 4 and 5). Increasing the temperature from 75 °C to 90 °C accelerated the reaction, providing 76% yield of 2b in 3 h (entry 5). Based on these results, acetic acid was selected as an optimal solvent for further exploration.
| Entry | Solvent(s) | T/°C | Time/h | Results |
|---|---|---|---|---|
| a Isolated yield of 2b; NR = no reaction. | ||||
| 1 | HCO2H | 25 | 48 | NR |
| 2 | HCO2H | 75 | 48 | NR |
| 3 | AcOH | 25 | 48 | NR |
| 4 | AcOH | 75 | 20 | 60%a |
| 5 | AcOH | 90 | 3 | 76%a |
| 6 | EtCO2H | 25 | 48 | NR |
| 7 | EtCO2H | 75 | 48 | NR |
| 8 | CF3CO2H | 25 | 48 | Complicated |
| 9 | 1 equiv. CF3CO2H in 1,2-DCE | 25 | 48 | NR |
| 10 | 1 equiv. CF3CO2H in 1,2-DCE | 75 | 40 | 35%a |
| 11 | 1 equiv. CF3CO2H in MeCN | 25 | 48 | NR |
| 12 | 1 equiv. CF3CO2H in MeCN | 75 | 40 | Complicated |
| 13 | 1 equiv. TfOH in 1,2-DCE | 25 | 48 | NR |
| 14 | 1 equiv. TfOH in 1,2-DCE | 75 | 40 | 30%a |
| 15 | 1 equiv. TfOH in MeCN | 25 | 48 | NR |
| 16 | 1 equiv. TfOH in MeCN | 75 | 40 | Complicated |
Under the optimized reaction conditions, a number of o-alkynylbenzaldehydes were examined having electron-withdrawing or alkyl substituent(s) on their benzene ring A, including halogen, carboxylate, nitro, cyano, trifluoromethyl and tert-butyl groups (Table 2). These substrates were all found to afford the corresponding dimeric products in various isolated yields (entries 2–8). The reaction of 1h (R1 = 5-CN, entry 7) gave only a 30% yield of the dimer 2h, owing to partial hydrolysis of the cyano group under the strongly acidic conditions. Three substrates having a single stronger electron-withdrawing group could undergo dimerization at room temperature with reactions reaching completion in relatively longer times (entries 3–5). Altering the R2 substituent on the phenyl ring on the opposite side of the acetylene functionality did not significantly change the course of the reactions (entries 11–15). To examine the electronic effects on dimerization caused by substituent(s) on the benzene ring A (Fig. 2), a substrate (1j, entry 9) having both an electron-donating group (5-OMe) and an electron-withdrawing group (4-NO2) was employed. The reaction gave the expected dimer 2j in a relatively low yield. In addition, the substrate 1k having a tert-butyl (a weakly electron-donating group) on the benzene ring also followed a similar pathway to afford the dimer 2k (entry 10).
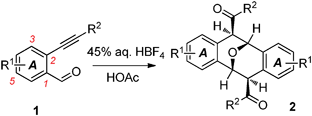
| Entry | 1 | T/°C | Time/h | 2 (%)a |
|---|---|---|---|---|
| a Isolated yield. | ||||
| 1 | 1b (R1 = H; R2 = Ph) | 90 | 3 | 2b (76%) |
| 2 | 1c (R1 = 5-Cl; R2 = Ph) | 100 | 24 | 2c (73%) |
| 3 | 1d (R1 = 6-F; R2 = Ph) | 25 | 48 | 2d (65%) |
| 4 | 1e (R1 = 5-CO2Me; R2 = Ph) | 25 | 48 | 2e (63%) |
| 5 | 1f (R1 = 5-NO2; R2 = Ph) | 25 | 48 | 2f (67%) |
| 6 | 1g (R1 = 3,5-di-Cl; R2 = Ph) | 100 | 60 | 2g (70%) |
| 7 | 1h (R1 = 5-CN; R2 = Ph) | 60 | 12 | 2h (30%) |
| 8 | 1i (R1 = 5-CF3; R2 = Ph) | 80 | 26 | 2i (72%) |
| 9 | 1j (R1 = 4-NO2/5-OMe; R2 = Ph) | 80 | 11 | 2j (20%) |
| 10 | 1k (R1 = 5-tBu; R2 = Ph) | 70 | 29 | 2k (60%) |
| 11 | 1l (R1= H; R2 = 4-ClC6H4) | 80 | 15 | 2l (80%) |
| 12 | 1m (R1 = H; R2 = 4-FC6H4) | 80 | 12 | 2m (82%) |
| 13 | 1n (R1 = H; R2 = 4-tBuC6H4) | 80 | 5 | 2n (67%) |
| 14 | 1o (R1 = H; R2 = 4-MeC6H4) | 80 | 23 | 2o (87%) |
| 15 | 1p (R1 = 5-CO2Me; R2 = 4-tBuC6H4) | 60 | 8 | 2p (65%) |
A comprehensive analysis of the results from this work and our previous study10 shows that only those o-alkynylbenzaldehydes bearing stronger electron-donating group(s) on their benzene ring A could be easily converted into the corresponding stable isochromenylium salts by treatment with tetrafluoroboric acid. Single crystal and layer-to-layer structures of ICTB 1a10 indicate that its stability benefits from the formation of multiple three-dimensional non-bonding interactions including π–π stacking, which are heavily dependent on the phenyl ring A having electron-donating group(s). Without sufficiently strong electron-donating group(s), the phenyl ring A could not serve as a good electron-rich π system capable of stabilizing the electron-deficient oxonium Hückel aromatic system in the ICTBs. Unfortunately, because the substrates 1b–1p examined in our current work (Table 2) lack such structural features, they do not favor formation of the corresponding stable ICTBs in the presence of 45% aq. HBF4 in acetic acid. Instead of intramolecular attack by the internal aldehyde carbonyl (leading to the corresponding ICTBs), water might serve as an alternative external nucleophile when the carbon–carbon triple bond of o-alkynylbenzaldehyde 1 is exposed to strong Brønsted acid conditions. Accordingly, the hydrolysis products derived from the o-alkynylbenzaldehydes should participate in the dimerization process.
To verify this possibility, we prepared two hydrolysis products (1q and 1s) from o-alkynylbenzaldehydes 1b and 1a, respectively (Scheme 1).12 Heating the dicarbonyl compound 1s with 45% aq. HBF4 in acetic acid at 75 °C afforded the corresponding ICTB 2a in 3 h in 73% yield; while the corresponding reaction of 1q did not provide any of the expected dimer or ICTB salt. Unexpectedly, reaction of a mixture of 1s (1 equiv.) and 1b (1 equiv.) resulted in smooth dimerization under the same conditions. However, the combination of 1a and 1s still afforded the corresponding ICTB 2a.
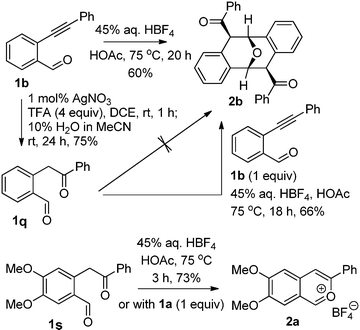 | ||
| Scheme 1 Examination of the proposed intermediates 1q and 1s. | ||
Based on the above evidence, using the conversion of 1b into Kagan's ether 2b as a representative example, a possible mechanism can be proposed to explain the Brønsted acid-promoted dimerization of o-alkynylbenzaldehydes (Fig. 3). Initially, the acetylene functionality of 1b is protonated under the acidic reaction conditions. This theoretically offers a competitive opportunity for two different attacks from oxygen functionalities. One attack involves intramolecular addition by the internal carbonyl (leading to the oxonium, ICTB), while the other attack is the simple hydrolysis by water. As discussed above, the ICTB pathway is unfavorable for substrates lacking strong electron-donating substituent(s) on the benzene ring A. Under these circumstances, a cross aldol reaction can take place between the enol B1, derived from the hydrolysis product A1, and the second molecule of aldehyde 1b. The resulting syn-aldol adduct C1 is quickly converted into the semi-acetal D1. Under acidic conditions, intramolecular C-glycosylation by the acetylene and simultaneous hydrolysis affords the final Kagan's ether 2b.
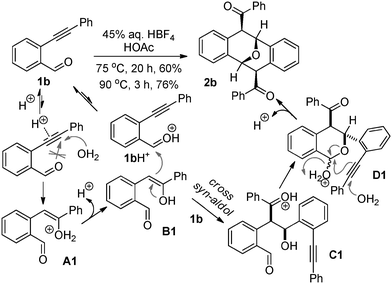 | ||
| Fig. 3 A proposed pathway for the dimerization of 1b. | ||
The newly discovered Brønsted acid-promoted dimerization of o-alkynylbenzaldehyde provides convenient one-step access to a variety of functionalized Kagan's ethers. However, limitations in the scope of substrates imposed by mechanistic considerations (Fig. 3), mean that only acyl groups can be directly introduced into the C4 and C8 positions of Kagan's ethers. To achieve greater diversity, we examined transformation of the obtained products 2 to other Kagan's ethers (Scheme 2). For example, Baeyer–Villiger oxidation of 2b with 2,2,2-trifluoroperoxyacetic acid13 afforded the dibenzoate derivative 3 at room temperature. Subsequent hydrolysis of dibenzoate 3 with K2CO3 in methanol afforded the diol analogue 4. Removal of the two hydroxyls of diol 4 could be accomplished by acetylation followed by hydrogenolysis, to afford the basic structure of Kagan's ether 6.2
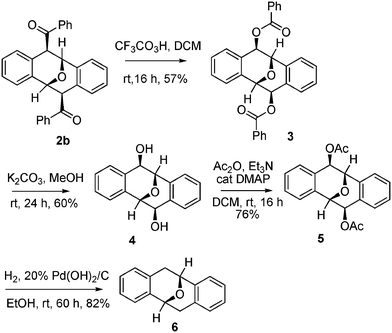 | ||
| Scheme 2 Transformations of 2b to various Kagan's ether derivatives. | ||
In conclusion, we report herein the Brønsted acid-promoted dimerization of o-alkynylbenzaldehydes via a cascade multistep process. The substrate scope was examined and potential mechanisms were proposed and discussed. The developed one-step methodology provides a convenient synthesis of symmetrical 2,3,6,7-dibenzo-9-oxabicyclo[3.3.1]nona-2,6-dienes (Kagan's ethers). In addition, the resulting Kagan's ethers have the advantage of being potentially transformed into additional Kagan's ethers.
References
- For reviews on Kagan's ether, see: (a) M. Harmata, M. Kahraman, S. Tyagarajan, C. L. Barnes and C. J. Welch, in Molecular Recognition and Inclusion, ed. A. W. Coleman, Kluwer, Dordrecht, 1998, p. 109 Search PubMed; (b) M. Harmata, Acc. Chem. Res., 2004, 37, 862 CrossRef CAS.
- (a) J. Kagan, S. Y. Chen, D. A. Agdeppa, W. H. Watson and V. Zabel, Tetrahedron Lett., 1977, 18, 4469 CrossRef; (b) J. Kagan, S.-y. Chen, D. A. Agdeppa Jr, W. H. Watson and V. Zabel, Tetrahedron Lett., 1978, 19, 4469 Search PubMed; (c) J. Kagan, D. A. Agdeppa, A. I. Chang, S. A. Chen, M. A. Harmata, B. Melnick, G. Patel, C. Poorker, S. P. Singh, W. H. Watson, J. S. Chen and V. Zabel, J. Org. Chem., 1981, 46, 2916–2930 CrossRef CAS.
- (a) B. Wünsch, M. Zott and G. Höfner, Arch. Pharm., 1992, 325, 733 CrossRef; (b) B. Wünsch, M. Zott and G. Höfner, Arch. Pharm., 1993, 326, 823 CrossRef; (c) C. Ketterer, S. Grimme, E. Weckert and B. Wünsch, Tetrahedron: Asymmetry, 2006, 17, 3046 CrossRef CAS.
- (a) C. Maurin, F. Bailly and P. Cortelle, Curr. Med. Chem., 2003, 10, 1795 CrossRef CAS; (b) R. Dupont, L. Jeanson, J.-F. Mouscadet and P. Cotelle, Bioorg. Med. Chem. Lett., 2001, 11, 3175 CrossRef CAS; (c) R. Dupont and P. Cotelle, Tetrahedron Lett., 1998, 39, 8457 CrossRef CAS.
- S. Bhunia, K.-C. Wang and R.-S. Liu, Angew. Chem., Int. Ed., 2008, 47, 5063 CrossRef CAS.
- M. Harmata and T. Murray, J. Org. Chem., 1989, 54, 3761 CrossRef CAS.
- (a) M. E. Jung, A. B. Mossman and M. A. Lyster, J. Org. Chem., 1978, 43, 3698 CrossRef CAS; (b) M. E. Jung and S. J. Miller, J. Am. Chem. Soc., 1981, 103, 1984 CrossRef CAS.
- M. Harmata, Y. Wu, M. Kahraman and C. J. Welch, Synth. Commun., 2001, 31, 3345 CrossRef CAS.
- M. Harmata and M. Kahraman, Tetrahedron Lett., 1999, 40, 4133 CrossRef CAS.
- Z.-L. Hu, W.-J. Qian, S. Wang, S.-z. Wang and Z.-J. Yao, Org. Lett., 2009, 11, 4676 CrossRef CAS.
- (a) Z.-L. Hu, W.-J. Qian, S. Wang, S.-z. Wang and Z.-J. Yao, J. Org. Chem., 2009, 74, 8787 CrossRef CAS; (b) Z.-L. Hu, W.-J. Qian and Z.-J. Yao, Sci. China: Chem., 2010, 53, 869 CrossRef CAS; (c) H.-Z. Liao, Z.-L. Hu, K. Cui, J. Jiao, Y. Qin and Z.-J. Yao, Synthesis, 2010, 3474 Search PubMed; (d) Z.-L. Hu, Z.-Y. Yang, S.-z. Wang and Z.-J. Yao, Chem.–Eur. J., 2011, 17, 1268 CrossRef CAS.
- (a) W.-J. Qian, W.-G. Wei, Y.-X. Zhang and Z.-J. Yao, J. Am. Chem. Soc., 2007, 129, 6400 CrossRef CAS; (b) J. Zhu, A. R. Germain and J. A. Porco Jr, Angew. Chem., Int. Ed., 2004, 43, 1239 CrossRef CAS.
- (a) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737 CrossRef CAS; (b) A. Bayer and V. Villiager, Ber. Dtsch. Chem. Ges., 1899, 32, 3625 CrossRef; (c) A. Bayer and V. Villiager, Ber. Dtsch. Chem. Ges., 1900, 33, 858 CrossRef; (d) A. Rassat and G. Ourisson, Bull. Soc. Chim. Fr., 1959, 1133 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of new compounds, NMR spectra of new compounds (PDF), and crystallographic data of compound 2b (CIF). CCDC 853872. For ESI and crystallographic data see DOI: 10.1039/c2ra20095c |
| ‡ This project is financially supported by the MOST 973 Program (2010CB833200), NSFC (21032002), SHMCST (08431903000), the Fundamental Research Funds for the Central Universities (No. 1082020502) and the State Key Laboratory of Phytochemistry and Plant Resources in West China. The authors thank Dr Terrence R. Burke, Jr. for kind assistance with language editing. |
| This journal is © The Royal Society of Chemistry 2012 |