Synthesis of 2-amino-imidazoles, purines, and benzoxazolamines through DIB oxidation†
Jean-Christophe
Andrez
*
Cardiome Pharma Corp, 6190 Agronomy Rd., 6th Floor, Vancouver, British Columbia, Canada V6T 1Z3. E-mail: jandrez@cardiome.com; Tel: 604-677-6905 (ext112)
First published on 24th February 2012
Abstract
A wide range of fused 2-amino-imidazoles and 2-amino-benzoxazoles can be prepared in one-pot under very mild conditions from 1,2-diamino-arenes or 2-amino-phenols and Vilsmeier reagents. The oxidation proceeds using diacetoxy-iodobenzene (DIB) and is tolerant to different functionalities.
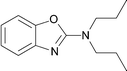
Fused arene-imidazoles are of significant importance to medicinal chemistry. Of particular interest are the benzimidazole-containing compounds, with purine and xanthine derivatives showing applications as inducers of interferon and lineage-committed cell differentiation, melanin-concentrating hormone receptors 1 (MCH R1) and transient receptor potential vanilloid 1 (TRPV1) receptors, ligands of corticotropin-releasing hormone receptors, and as inhibitors of heat shock protein 90 (HSP90), Src kinase, p38a mitogen-activated protein (MAP) kinase, sulfotransferases, phosphodiesterases, and cyclin-dependent kinases (CDKs).1 Benzoxazoles are also important targets in drug discovery as human immunodeficiency virus type 1 (HIV-1) protease inhibitors, 5-hydroxytryptamine type 3 (5-HT3) agonist, leukotriene B4 (LTB4) inhibitors, and serotonergic and histaminergic receptor antagonists.2
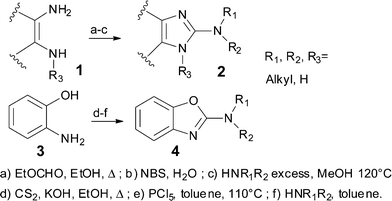
Whereas the syntheses of simple 2-alkyl-arene-imidazoles or 2-alkyl-benzoxazoles are well established,3 the preparation of their 2-amino equivalents relies mostly on long syntheses and harsh reaction conditions4 (Scheme 1). Interestingly, although these molecules are extensively studied in the pharmaceutical industry for the preparation of drug libraries, no general method exists in the literature that would allow for both a high level of diversity and efficiency.5 Recently, other methods using metal catalysed ring closure have been successfully employed,6 but suffer from the use of toxic materials, strong bases, high temperatures, and the need for preparation and purification of the starting materials.
 | ||
| Scheme 1 Traditional formation of 2-amino-imidazoles and benzoxazolamines. | ||
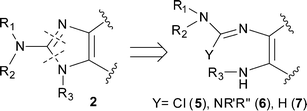
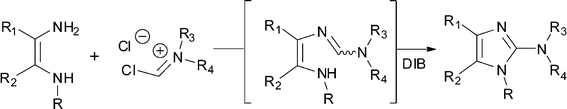
This paper describes a new one-step general procedure for the synthesis of fused 2-amino-imidazoles 2 and 2-benzoxazolamines 4 that tolerates a wide variety of functionalities. We were interested in accessing compounds of type 2 bearing a variety of substituents at R1, R2, and R3 (Fig. 1). Cyclisations of compounds bearing a guanidine 6 or a carbamimidic chloride 5 are documented.7,8 However, these reactions are also usually done with simple alkyl groups (R1 = R2 = Me, H) and electron rich heteroaromatic rings.9
 | ||
| Fig. 1 Disconnection of the 2-amino-imidazole ring. | ||
Initial attempts to use literature procedures to prepare compounds of general structure 2 from 1,2-diamino compounds 1 and 2-chloro-1,1,3,3-tetraalkylformamidinium chloride7 or phosgene iminium chloride8 could only provide the simple N,N-dimethyl-amino-imidazole derivatives. Other attempts to introduce larger substituents on the exocyclic amine failed to produce the cyclized product, even under forcing conditions. We envisioned a new avenue to these compounds involving an oxidative cyclisation of compound 7.
Gratefully, when reacting the readily available Vilsmeier reagent 9 (1.2 equiv.) with the 5,6-diamino-1,3-dimethyluracil 8, the formamidine intermediate 10 formed within minutes‡ and subsequent addition of diacetoxy-iodobenzene (DIB, 1 equiv.) triggered the cyclisation and oxidation to give xanthine 14 in 97% yield (Scheme 2).
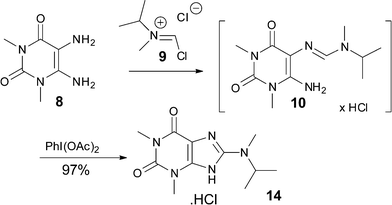 | ||
| Scheme 2 2-Amino-imidazole formation. | ||
The same chemistry can be used to form other fused 2-amino-imidazole ring systems with good to excellent yields. Electron deficient systems such as uracils or maleonitriles (precursors for purine derivatives) react readily and efficiently to give the fused imidazoles (Table 1, entries 1, 2, 4, and 6; 82–97% yield). It was interesting to observe that a benzyl-carbamate (Table 1, entry 2) survived during the course of this reaction. Reactions with more electron rich benzene diamine compounds (Table 1, entries 3 and 5) gave the fused imidazoles with only a slight erosion of the yield (65–73% yield). It was noticed that during the reaction to produce compound 15, the corresponding N-methyl-benzimidazole was formed to a small extent (∼10%).§ Whether this decomposition occurred during the formation of the formamidine or after the addition of DIB is unknown.¶ When the reaction was performed at a lower temperature (−78 °C)|| in order to decrease the amount of this side product, an intractable mixture was formed. We suspect that oxidation of the benzene ring may be a competitive process under these conditions, similar to that of aniline derivatives.10
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Producta | Yieldb | Entry | Producta | Yieldb | ||
| a Reactions were run in DCM–acetonitrile solution for imidazole syntheses, and DCM–1,4-dioxane solution for oxazole syntheses at ca. 0.2 M with 1.2 equiv. of the Vilsmeier reagent and 1.1 equiv. of DIB at 20 °C. For imidazole syntheses, water was added before the addition of DIB. For oxazole syntheses, aqueous NaOH was added before the addition of DIB. For most substrates, reactions were complete in less than 10 min except for entry 4 in which no water was added after the formation of the formamidine. It resulted in a heterogeneous reaction with DIB, and consequently a longer reaction time was required for the reaction to go to completion (10 h). b Isolated yield. c Isolated as the HCl salt. | |||||||
| 1 |
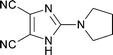
|
11 | 86 | 6 |
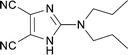
|
16 | 86 |
| 2 |
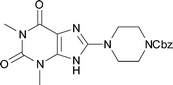
|
12 | 87 | 7 |
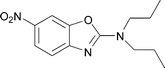
|
17 | 67 |
| 3 |
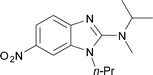
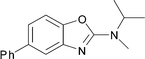
|
13 | 73 | 8 |
|
18 | 81 |
| 4 |
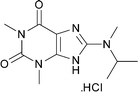
|
14 | 97c | 9 |
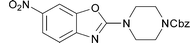
|
19 | 56 |
| 5 |
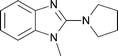
|
15 | 65 | 10 |
|
20 | 74 |
DIB was the oxidant of choice, but oxone was also effective in promoting the imidazole formation, albeit in lower yield. Perbenzoic acid, TEMPO, and other inorganic oxidants (NaBrO3, KIO3, KNO3) failed to give the 2-amino-imidazoles.
The reaction also succeeds with 2-amino-phenols giving access to valuable benzoxazolamines in a moderate to good yield (Table 1, entries 7, 8, 9, and 10). In these syntheses, sodium hydroxide was added to the reaction mixture before the addition of DIB to prevent the degradation of acid sensitive oxazoles. Interestingly, when reacting 2-amino-4-chlorophenol 21 under the standard reaction conditions, the expected benzoxazolamine was detected in only 23% yield by LCMS and compound 22 was isolated as the major product in 56% yield (Scheme 3).**
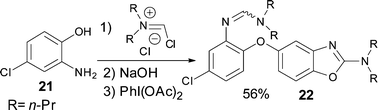 | ||
| Scheme 3 SNAr on activated phenol 21. | ||
This multi-component reaction (MCR) as well as the versatility of the DIB reaction towards different substrates can be very useful for the discovery of biologically active molecules via a diversity-oriented synthesis (DOS) approach.
A tentative mechanism for the formation of compound 22 is outlined in Scheme 4. After formation of formamidine 23,†† we suppose that the addition of sodium hydroxide and DIB triggers the oxidation of the aromatic ring11 to give the stabilized carbocation 24, which may either undergo direct conversion to the benzoxazolamine or be intercepted by the phenolate derived from 23 through an addition–elimination process to give 25.12 A final deprotonation–cyclisation can provide the observed compound 22.13
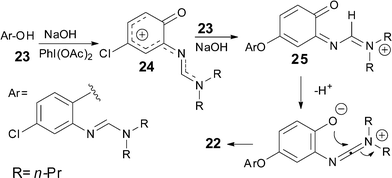 | ||
| Scheme 4 Mechanistic hypothesis for formation of 22. | ||
Whether this observation has general mechanistic implications for this methodology is so far unknown, but it is interesting to note that the formation of compound 18, which would involve the most stabilized carbocation intermediate, gave the best yield. We can infer that the 4-phenyl group increases the life-time of the intermediate by decreasing its electrophilicity, and therefore participates to enhance the selectivity of the reaction.
Conclusions
In conclusion, a practical and new method for the synthesis of fused imidazoles and benzoxazolamines is now available. This transformation provides milder reaction conditions than previous reports and allows the assembly of those compounds in a one-step oxidative-cyclisation process. The starting materials are either commercially available or easily synthesized by known means. The method has proven useful for the preparation of analogs in a multigram scale and will make the preparation of other more elaborate fused imidazole rings simpler and more readily accessible. Moreover, an unexpected and interesting outcome occurred with the reaction of 4-chloro-2-aminophenol which could allow for the introduction of different kinds of nucleophiles at the 4-position of the benzoxazolamine. Preparation of other heterocycles using this methodology, investigation of the mechanism of 4-functionalized benzoxazolamines, and its application to the one-pot synthesis of other 4-substituted benzoxazolamines is currently being investigated in the laboratory and will be disclosed in due course.Acknowledgements
We thank Dr A. Eniade, Cardiome Pharma Corp, for the preliminary work. We are also grateful to B.M.T. and I. Cho from the Department of Chemistry of Simon Fraser University, and all the scientists of the chemistry department of Cardiome Pharma Corp for their helpful discussions.References
- For examples of arene-imidazoles in drug discovery, see: (a) M. Legraverend and D. S. Grierson, Bioorg. Med. Chem., 2006, 14, 3987 CrossRef CAS and references cited therein; (b) K. A. Jacobson, P. J. M. Van Galen and M. Williams, J. Med. Chem., 1992, 35, 407 CrossRef CAS; (c) V. I. Ognyanov, C. Balan and A. W. Bannon, J. Med. Chem., 2006, 49, 3719 CrossRef CAS; (d) A. J. Carpenter, K. A. Al-Barazanji, K. K. Barvian, M. J. Bishop, C. S. Britt, J. P. Cooper, A. S. Goetz, M. K. Grizzle, D. L. Hertzog, D. M. Ignar, R. O. Morgan, G. E. Peckham, J. D. Speake and W. R. Swain, Bioorg. Med. Chem. Lett., 2006, 16, 4994 CrossRef CAS.
- For examples of benzoxazoles in drug discovery, see: (a) L. N. G. Surlereaux, H. A. de Kock and P. B. T. P. Wigerinck, J. Med. Chem., 2005, 48, 1965 CrossRef; (b) D. R. Buckle, B. C. C. Cantello, M. A. Cawthorne, P. J. Coyle, D. K. Dean, A. Faller, D. Haigh, R. M. Hindley, L. J. Jefcott, C. A. Lister, I. L. Pinto, H. K. Rami, D. G. Smith and S. A. Smith, Bioorg. Med. Chem. Lett., 1996, 17, 2127 CrossRef; (c) Y. Sato, M. Yamada, S. Yoshida, T. Soneda, M. Ishikawa, T. Nizato, K. Suzuki and F. Konno, J. Med. Chem., 1998, 41, 3015 CrossRef CAS; (d) G. P. Roth, J. J. Landi, A. M. Salvagno and H. Muller-Botticher, Org. Process Res. Dev., 1997, 1, 331 CrossRef CAS; (e) E. S. Lazer, C. K. Miao, H.-C. Wong, R. Sorcek, D. M. Spero, A. Gilman, K. Pal, M. Behnke, A. G. Graham, J. M. Watrous, C. A. Homon, N. Juergen, A. Shah, Y. Guindon, P. R. Farina and J. Adams, J. Med. Chem., 1994, 37, 913 CrossRef CAS; (f) H. Razavi, S. K. Palaninathan, E. T. Powers, R. L. Wiseman, H. E. Purkey, N. N. Mohamedmohaideen, S. Deechongkit, K. P. Chiang, M. T. A. Dendle, J. C. Sacchettini and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 2758 CrossRef CAS; (g) K. G. Liu, J. R. Lo, T. A. Comery, G. M. Zhang, J. Y. Zhang, D. M. Kowal, D. L. Smith, L. Di, E. H. Kerns, L. E. Schechter and A. J. Robichaud, Bioorg. Med. Chem. Lett., 2009, 19, 1115 CrossRef CAS.
- For a general overview to prepare benzoxazoles, see: (a) G. V. Boyd, in Sciences of Synthesis: Houben-Weyl Methods of Molecular Transformations, ed. E. Schaumann, Thieme, Stuttgart, 2002, vol. 11, pp. 481. Search PubMedFor a general overview to prepare arene-imidazoles, see: (b) L.-H. Du and Y.-G. Wang, Synlett, 2007, 5, 675 Search PubMed and references cited therein.
- For amino-imidazoles synthesis, see: (a) C. Jin, J. P. Burgess, K. S. Rehder and G. A. Brine, Synthesis Stuttgart, 2007, 219 CAS; (b) A. Orjales, R. Mosquera, L. Labeaga and R. Rodes, J. Med. Chem., 1996, 40, 586 CrossRef; (c) I. Feoktistov, E. M. Garland, A. E. Goldstein, D. Zeng, L. Belardinelli, J. N. Wells and I. Biaggioni, Biochem. Pharmacol., 2001, 62, 1163 CrossRef CASFor benzoxazolamines synthesis, see (d) S. Yoshida, T. Watanabe and Y. Sato, Bioorg. Med. Chem., 2007, 15, 3515 CrossRef CAS.
- (a) J. Y. Hwang and Y.-D. Gong, J. Comb. Chem., 2006, 8, 297 CrossRef CAS; (b) C. H. Soh, W. K. Chui and Y. Lam, J. Comb. Chem., 2008, 10, 118 CrossRef CAS.
- (a) B. G. Szczepankiewicz, J. J. Rohde and R. K. Kurukulasuriya, Org. Lett., 2005, 7, 1833 CrossRef CAS; (b) C. T. Brain and S. A. Brunton, Tetrahedron Lett., 2002, 43, 1893 CrossRef CAS.
- K. Ohno, W. Ishida, K. Kamata, K. Oda and M. Machida, Heterocycles, 2003, 59, 317 CrossRef CAS.
- C. Papamicaël, G. Dupas, G. Quéguiner and J. Bourguignon, Heterocycles, 1998, 47, 991 CrossRef.
- S. Weiss, H. Michaud, H. Prietzel and H. Krommer, Angew. Chem., Int. Ed., 1973, 12, 841 CrossRef.
- (a) S. Akai, N. Kawashita, N. Morita, Y. Nakamura, K. Iio and Y. Kita, Heterocycles, 2002, 58, 75 CrossRef CAS; (b) P. V. Zawada, S. C. Banfield and M. A. Kerr, Synlett, 2003, 971–974 CAS.
- (a) J.-C. Andrez, M.-A. Giroux, J. Lucien and S. Canesi, Org. Lett., 2010, 12, 4368 CrossRef CAS; (b) S. Akai, N. Kawashita, N. Morita, Y. Nakamura, K. Lio and Y. Kita, Heterocycles, 2002, 58, 75 CrossRef CAS; (c) S. Desjardins, J.-C. Andrez and S. Canesi, Org. Lett., 2011, 13, 3406 CrossRef CAS.
- For examples of 1,4 addition on o-azaquinones see: (a) K. C. Nicolaou, K. Sugita, P. S. Baran and Y. L. Zhong, J. Am. Chem. Soc., 2002, 10, 2221 CrossRef and references therein. For other examples of hypervalent iodine-induced nucleophilic substitution on aromatics see (b) Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 9, 3684 CrossRef.
- (a) We expect the iminium ion 25 to be easily deprotonated at the C-sp2 carbon either through a [1,5]hydrogen shift or by an external base. For discussion about deprotonation of related compounds see: D. B. Dupre and J. L. Wong, J. Phys. Chem. A, 2007, 111, 2172 CrossRef CAS; (b) S. Brandange and L. Lindblom, Chem. Commun., 1979, 650 RSC.
Footnotes |
| † Electronic Supplementary Information (ESI) available: See DOI: 10.1039/c2ra20101a/ |
| ‡ The formamidine can be isolated and characterized. A. Larizza, G. Brancaccio and G. Lettieri, J. Org. Chem., 1964, 29, 3697. In this study, the formamidine was not isolated but observed by LCMS. Both the formamidine and the corresponding cyclised N,N,N-orthoester may form under those conditions. LCMS analysis can not discern between either of these two forms. |
| § LCMS analysis of the reaction mixture after DIB oxidation showed the formation of this compound in ∼10% area. |
| ¶ Nonetheless, heating the formamidine intermediate at 50 °C for 15 min caused the complete conversion to the N-methyl-benzimidazole. |
| || After formation of the formamidine intermediate and the addition of water and DIB, the reaction mixture was allowed to warm up slowly to room temperature (1 h). |
| ** Full 2D NMR spectral analysis (600 MHz) confirmed the structure of this compound. See ESI.† |
| †† LCMS analysis showed the complete formation of the formamidine or the cyclised N,N,N-orthoester. |
| This journal is © The Royal Society of Chemistry 2012 |