RuCl2(PPh3)3-catalyzed chemoselective hydrogenation of β, δ-diketo acid derivatives at the β-carbonyls†
Wan-Fang
Li
a,
Xiao-Min
Xie
a,
Xiao-Ming
Tao
a,
Xin
Ma
a,
Wei-Zheng
Fan
a,
Xiao-Ming
Li
a and
Zhao-Guo
Zhang
*ab
aSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China
bShanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai 200032, China. E-mail: zhaoguo@sjtu.edu.cn
First published on 8th March 2012
Abstract
Chemoselective reduction of the β-carbonyls in β, δ-diketo acid derivatives was achieved through RuCl2(PPh3)3 catalyzed homogeneous hydrogenation. Tetrahydrofuran (THF) played a key role in the chemoselectivity control. This atom-economical protocol provided β-hydroxy-δ-keto esters and amides as useful intermediates in good to excellent yields.
The reduction of ketones to alcohols is a very fundamental organic transformation,1 which has been routinely accomplished by various alumino- or borohydrides.2 However, large-scale use of these stoichiometric reagents often suffers from safety concerns, high cost and tedious work-up. Besides, common reductants like NaBH4 and LiAlH4 show hardly any chemoselectivity for poly-carbonyl substrates,3 especially when the carbonyls are in similar steric and electronic environment.4
For example, the selective reduction of the β, δ-diketo acid derivatives at the desired position is important but difficult to control. As illustrated in Scheme 1, metal hydrides and their modifiers tend to reduce both β- and δ- carbonyls and semi-reduction products can rarely be attained except for a few sterically hindered substrates.5 Even many ketoreductases were unable to give an ideal chemoselectivity.6
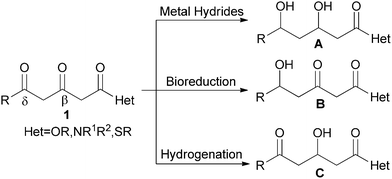 | ||
| Scheme 1 Various methods for reduction of 1. | ||
Several recently-developed biocatalysts reduced the δ-carbonyls with spectacular chemo- and enantioselectivity,6c,7 although few of them can reduce β-carbonyls equally well.6c Moreover, the high costs and narrow substrate scope limited their applications. Until now, no practical reduction methods with good β-selectivity have been reported.
Homogeneous hydrogenation has been increasingly employed in the reduction of unsaturated functionalities due to its unique merits.8 Earlier attempts on the β-selective hydrogenation of β, δ-diketo esters failed because the initial hydrogenation products easily underwent further hydrogenation in alcohols.9 In 1999, Carpentier et al.9a implemented a β-selective hydrogenation of methyl 3,5-dioxohexanoate with a Ru-BINAP system under the specified conditions (20 to 50 °C, 100 atm of H2, CH2Cl2). However, the reactions must be carefully monitored by GLC during the course to avoid over hydrogenation.
In our earlier studies on the asymmetric hydrogenation of 3-oxoglutaric acid derivatives,10 we discovered that a coordinative solvent like THF helped to modulate the coordination ability of carbonyls in β-keto esters and β-keto amides. The latter were hydrogenated much more quickly than the former.
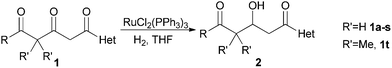
Based on these conclusions, we tried the hydrogenation of various β, δ-diketo acid derivatives in THF, which was an ineffective solvent for the catalytic hydrogenation of many functionalized ketones.11 To our pleasure, good to excellent yields were obtained with RuCl2(PPh3)3 as the catalyst under moderate H2 pressure (20 bar).
Simple optimization of the reaction conditions with 1b and 1m was performed. It took 30 h for 90% conversion of 1m at 50 °C under 5 bar of H2 with S/C = 100. At 90 °C, the reaction became complex for esters but was still good for amides. Thus we performed hydrogenation reactions at 70 °C and at 20 bar of H2. Under such relatively mild conditions, all the tested substrates converted to the desired products in 8 to 15 h. A prolonged reaction time (especially for the amides) after full conversion (8 h for most amides) lead to no over reduction. The resistance to further hydrogenation of the β-hydroxy amides was illustrated by the fact that 2m remained untouched when subjected to harsher conditions (1% mmol of catalyst, 20 bar of H2, 95 °C, 8 h). Therefore, cautious monitoring of the hydrogenation process is unnecessary.
As listed in Table 1, the esters 1a–e gave the desired products in good yields. The (hetero)aryl substituted esters (entries 3–5, Table 1) gave somewhat higher yields than the alkyl substituted esters (entries 1–2, Table 1), as the latter was partially converted to some byproducts.9a Various amides, especially the aryl substituted ones, were cleanly hydrogenated in almost quantitative yields. Diethyl amides reacted faster than tert-butyl esters. For example, 1m, 1n and 1o underwent full conversion in 8 h while 1b and 1d did not. This was also consistent with the hydrogenation rate sequence reported by Brückner et al.12 Electron withdrawing or donating substituents on different positions of phenyl rings had little influence on the reactions and the heteroatoms, such as sulfur in the thiophene did not affect the activity of the catalyst in this reaction (entry 17, Table 1).13 Substrates with geminal dimethyl substituents on the γ-methylene (1t) can also be hydrogenated smoothly. The asymmetric version of this type of substrates were used in the synthesis of epothilone and its congeners.14
| Entry | 1 | R | Het | Yield(%)b |
|---|---|---|---|---|
| a All reactions were carried out with 1 mmol of substrate in 5 mL of THF at 70 °C under 20 bar of H2 for 8–15 h. S/C = 200. Conversion = 100%. 8 h is enough for most amide substrates. b Isolated yields. | ||||
| 1 | 1a | Me | OMe | 85 |
| 2 | 1b | Me | OtBu | 86 |
| 3 | 1c | Ph | OtBu | 87 |
| 4 | 1d | 2-Naphthyl | OtBu | 90 |
| 5 | 1e | 2-Furanyl | OtBu | 92 |
| 6 | 1f | Et | NHtBu | 95 |
| 7 | 1g | Cy | NHtBu | 91 |
| 8 | 1h | Me | N-Morpholinyl | 91 |
| 9 | 1i | tBu | N-Morpholinyl | 93 |
| 10 | 1j | C6H5 | N-Morpholinyl | 95 |
| 11 | 1k | Me | NBn2 | 94 |
| 12 | 1l | Ph(CH2)2 | NEt2 | 95 |
| 13 | 1m | C6H5 | NEt2 | 96 |
| 14 | 1n | 2-MeC6H5 | NEt2 | 94 |
| 15 | 1o | 3-ClC6H5 | NEt2 | 96 |
| 16 | 1p | 4-MeOC6H5 | NEt2 | 96 |
| 17 | 1q | 4-CF3C6H5 | NEt2 | 95 |
| 18 | 1r | 1-Naphthyl | NEt2 | 95 |
| 19 | 1s | 2-Thienyl | NEt2 | 96 |
| 20 | 1t | Et | NEt2 | 91 |
To investigate which carbonyl was the directing group in the β-selective hydrogenation, competitive reaction tests with a, b and c were performed (Scheme 2). Both β-keto ester (a) and β-diketone (b) were inert in the same reaction conditions, while the β-keto amide (c) can be hydrogenated smoothly to give the hydroxy product. One can envision that the 1,3-dicarbonyls in β-keto amides chelated with the ruthenium center, an essential step for the hydrogenation to proceed.15 In coordinative solvents like THF, which may act as a competitive ligand,16 the chelation ability of 1,3-dicarbonyl system in β-diketones and β-keto esters were comparably depleted. Intriguingly, the monofunctionalized ketones a and b were both inert to hydrogenation in THF, whereas 1a, a combined form of these two components was hydrogenated smoothly at the β-position. It was likely that the ruthenium coordinated to the enol forms of the substrates.9a As for the amide substrates, we suspected that the 1-carbonyls were the dominant directing groups, as depicted in Scheme 2, but further evidence is needed to determine if the coordination was related to the enol form at the β-carbon.
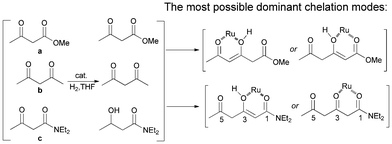 | ||
| Scheme 2 Proposed chelation modes in the hydrogenation reaction. | ||
In summary, we have developed a general method for the highly chemoselective reduction of the β, δ-diketo acid derivatives at the β-positions. The resultant products are very useful building blocks for organic synthesis. More arrestingly, this method employed the readily available RuCl2(PPh3)3 as the catalyst17 and inexpensive hydrogen as the green reductant. The asymmetric version of this reaction with chiral catalysis for simultaneous high chemo- and enantioselectivity is more challenging and is ongoing in our lab.
This work was financially supported by National Natural Science Foundation of China and Science and Technology Commission of Shanghai Municipality. We thank Johnson Matthey (Shanghai) Chemicals Limited for generous loan of precious metal catalysts.
References
- (a) N. Greeves, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, 1991, vol. 8, pp. 1–30 Search PubMed; (b) P. G. Andersson and I. J. Munslow, ed. Modern Reduction Methods, Wiley VCH, Weinheilm, 2008 Search PubMed.
- (a) H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567 CrossRef CAS; (b) J. Seyden-Pennein, Reductions by the Alumino- and Borohydrides in Organic Synthesis, Wiley-VCH, 1997 Search PubMed; (c) W. G. Brown, in Organic Reactions, John Wiley & Sons, Inc., 1951, vol. 6 Search PubMed.
- Z. Guo, Y. Chen, A. Goswami, R. L. Hanson and R. N. Patel, Tetrahedron: Asymmetry, 2006, 17, 1589 CrossRef CAS.
- B. M. Trost, Science, 1983, 219, 245 CAS.
- (a) G. B. Reddy, T. Minami, T. Hanamoto and T. Hiyama, J. Org. Chem., 1991, 56, 5752 CrossRef CAS; (b) T. Hiyama, G. B. Reddy, T. Minami and T. Hanamoto, Bull. Chem. Soc. Jpn., 1995, 68, 350 CrossRef CAS; (c) T. Hiyama, T. Minami and K. Takahashi, Bull. Chem. Soc. Jpn., 1995, 68, 364 CrossRef CAS.
- (a) M. Wolberg, I. A. Kaluzna, M. Müller and J. D. Stewart, Tetrahedron: Asymmetry, 2004, 15, 2825 CrossRef CAS; (b) R. Patel, A. Banerjee and L. Szarka, J. Am. Oil Chem. Soc., 1995, 72, 1247 CrossRef CAS; (c) R. N. Patel, A. Banerjee, C. G. McNamee, D. Brzozowski, R. L. Hanson and L. J. Szarka, Enzyme Microb. Technol., 1993, 15, 1014 CrossRef CAS.
- (a) M. Ernst, B. Kaup, M. Müller, S. Bringer-Meyer and H. Sahm, Appl. Microbiol. Biotechnol., 2005, 66, 629 CrossRef CAS; (b) S. Lüdeke, M. Richter and M. Müller, Adv. Synth. Catal., 2009, 351, 253 CrossRef; (c) M. Müller, Angew. Chem., Int. Ed., 2005, 44, 362 CrossRef; (d) M. Müller, M. Wolberg, T. Schubert and W. Hummel, Adv. Biochem. Engin./Biotechnol., 2005, 92, 261 Search PubMed; (e) M. Wolberg, W. Hummel, C. Wandrey and M. Müller, Angew. Chem., Int. Ed., 2000, 39, 4306 CrossRef CAS; (f) M. Wolberg, A. Ji, W. Hummel and M. Müller, Synthesis, 2001, 937 CrossRef CAS.
- (a) R. E. Harmon, S. K. Gupta and D. J. Brown, Chem. Rev., 1973, 73, 21 CrossRef CAS; (b) G. Dolcetti and N. W. Hoffman, Inorg. Chim. Acta, 1974, 9, 269 CrossRef CAS; (c) G. Shang, W. Li and X. Zhang, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Hoboken, New Jersey, 3rd edn, 2010 Search PubMed; (d) B. Cornils and W. A. Herrmann, ed. Applied Homogeneous Catalysis with Organometallic Compounds, Second, Completely Revised and Enlarged edn, Wiley-VCH, Weinheim, 2002 Search PubMed; (e) J. G. de Vries and C. J. Elsevier, ed., The Handbook of Homogeneous Hydrogenation, Wiley-VCHWeinheim, 2007 Search PubMed.
- (a) V. Blandin, J. F. Carpentier and A. Mortreux, Eur. J. Org. Chem., 1999, 3421 CrossRef CAS; (b) O. Labeeuw, C. Roche, P. Phansavath and J.-P. Genêt, Org. Lett., 2006, 9, 105 CrossRef.
- W. F. Li, X. Ma, W. Z. Fan, X. M. Tao, X. M. Li, X. M. Xie and Z. G. Zhang, Org. Lett., 2011, 13, 3876 CrossRef CAS.
- (a) A. Wolfson, I. F. J. Vankelecom, S. Geresh and P. A. Jacobs, J. Mol. Catal. A: Chem., 2003, 198, 39 CrossRef CAS; (b) E. Starodubtseva, O. Turova, M. Vinogradov, L. Gorshkova and V. Ferapontov, Russ. Chem. Bull., 2007, 56, 552 CrossRef CAS.
- (a) R. Kramer and R. Brückner, Angew. Chem., Int. Ed., 2007, 46, 6537 CrossRef CAS; (b) R. Kramer and R. Brückner, Chem.–Eur. J., 2007, 13, 9076 CrossRef CAS.
- The nitrogen atom in the pyridine ring inhibited the catalyst. For example, when R = 2-pyridinyl or 3-pyridinyl, the substrates became totally unreactive toward the catalytic reaction. A sharply different dark red solution was formed.
- (a) A. Rivkin, F. Yoshimura, A. E. Gabarda, Y. S. Cho, T.-C. Chou, H. Dong and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 10913 CrossRef CAS; (b) T. C. Chou, H. Dong, A. Rivkin, F. Yoshimura, A. E. Gabarda, Y. S. Cho, W. P. Tong and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 4761 CrossRef.
- (a) R. Noyori, M. Kitamura and T. Ohkuma, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5356 CrossRef CAS; (b) In Organotransition metal chemistry - from bonding to catalysis, ed. J. F. Hartwig, University Science Books, Sausalito, California, 2010, pp. 599 Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- Other complexes with readily available ligands such as rac-BINAP could also furnish the reaction with comparable results under the same conditions. RuCl2(PPh3)3 was chosen for cost consideration.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20114c/ |
| This journal is © The Royal Society of Chemistry 2012 |