Improved electrochemical performances of nanocrystalline Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode material for Li-ion batteries
Wei
He
a,
Jiangfeng
Qian
b,
Yuliang
Cao
*a,
Xinping
Ai
b and
Hanxi
Yang
*b
aCollege of Chemistry and Molecular Science, Wuhan University, Wuhan, 430072, P. R. China. E-mail: ylcao@whu.edu.cn; Tel: 86-027-68754526
bHubei Key Lab. of Electrochemical Power Sources, Wuhan University, Wuhan, 430072, P. R. China. E-mail: hxyang@whu.edu.cn
First published on 26th January 2012
Abstract
Layered Li[Li0.2Co0.13Ni0.13Mn0.54]O2 nanoparticles were synthesized by a simple polymer-pyrolysis method and then coated with 3 wt% Al2O3 to form a ∼4 nm thick protective skin. The Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 electrode demonstrates a high initial coulombic efficiency of 96.1%, a large reversible capacity of ∼311 mAh g−1, and a good cyclability with 83.8% capacity retention after 70 cycles. Particularly, this material can deliver a quite high capacity of ∼239 mAh g−1 at a high rate of 400 mA g−1. This superior electrochemical performance results from the well-crystallized nanocores and effective surface modification of the material. The former provides a short diffusion path and fast transport channels for lithium ion insertion/extraction reactions and the latter restrains the elimination of oxide ion vacancies and metal ion rearrangement during charge–discharge cycling. Due to their simplicity and applicability, the synthetic method along with the surface modification technique is easily adopted to make high performance xLi2MnO3·(1 − x)LiMO2 materials for practical battery applications.
1. Introduction
Li-ion batteries are considered to be a promising electric storage technology for upcoming electric vehicles (EV) and renewable-energy power stations, however, they have insufficient energy storage capacities which mostly arise from the Li-host cathodes, still a great obstacle for these high capacity applications.1–5 To solve this problem, a vast array of high capacity Li-storage cathode materials has been explored in the past two decades, among which layered Li2MnO3–stablized LiMO2 (M = Mn, Ni, Co, etc.) compounds (nominal xLi2MnO3·(1 − x)LiMO2 solid solutions) have received particular attention in recent years because of their realizable high capacity of ∼300 mAh g−1, nearly twice as high as presently commercialized LiCoO2 and LiFePO4 cathodes.6–9Despite their exceptional high capacity and low cost, the xLi2MnO3·(1 − x)LiMO2 materials suffer from two major disadvantages: low initial coulombic efficiency and poor high rate capability, which bring about great difficulties for practical applications. Though there has been no definitive evidence presented so far, the large initial irreversible capacity loss is usually attributed to an irreversible removal of partial lithium as Li2O, along with an elimination of the oxygen vacancies from the crystal lattice produced during first charge, which leads to a reduction of the effective sites for accommodating the lithium ions in subsequent cycles.10,11 Similarly, several mechanisms, such as the formation of a thick solid-electrolyte interface (SEI) on the cathode surface and the frustrated diffusion of lithium ions in the rearranged lattice formed during the first charge, have been proposed to account for the low rate capability of the Li2MnO3·LiMO2 materials, but the rapid capacity fading of the materials with increased charge and discharge rate is not fully understood.12–15
One effective strategy to suppress the irreversible capacity is to modify the surface chemistry of the materials for retaining the oxygen ion vacancies in the lattice upon first charge. As demonstrated by several independent groups, the surface coating of metal oxides, phosphates and fluorides can substantially improve the initial discharge capacity and exhibit a greatly improved cyclability, possibly due to its strong ability to retain the oxide ion vacancies in the lattice at subsequent charge–discharge cycles.16–24
To improve the rate capability of the layered xLi2MnO3·(1 − x)LiMO2 materials, a number of research groups have recently adopted different synthetic methods to optimize the kinetically suitable morphologies and sizes for these materials.25,26 Usually, lowering the particle size can considerably enhance the high rate capability of these materials. It has been reported the nanowires of the layered xLi2MnO3·(1 − x)LiMO2 electrodes can still deliver a high capacity of > 200 mAh g−1 at a high rate of 5C.27
In this work, we adopted a polymer-pyrolysis method to prepare nanosized Li[Li0.2Co0.13Ni0.13Mn0.54]O2 (denoted in a two-component notation as 0.5 Li2MnO3·0.5 LiMn1/3Ni1/3Co1/3O2) and coated this material with Al2O3, so as to obtain a high capacity, coulombic efficiency and rate cathode material. Compared with a conventional co-precipitation method, the polymer-pyrolysis method developed in our group can easily produce layered xLi2MnO3·(1 − x)LiMO2 nanoparticles with homogeneously dispersed transition metal ions in the lattice and a uniform size distribution at 100–150 nanometres, which are expected to improve the capacity utilization and rate capability of this material.
2. Experimental
2.1. Materials and characterization
Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powders were prepared by a polymer-pyrolysis method using polyacrylates of Li+, Ni2+, Co2+ and Mn2+ as precursors. The copolymeric precursor was made by solution polymerization of the mixed aqueous solution of acrylic acid in the presence of LiOH, Ni(NO3)2, Co(NO3)2 and Mn(NO3)2, with (NH4)2S2O8 as an initiator. The experimental procedure was first to dissolve LiOH·H2O, Ni(NO3)2·6H2O, and Mn(NO3)2 (aqueous solution) in aqueous acrylic acid solution (acrylic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 70:30 wt%) under stirring, and then add a small amount of (NH4)2S2O8 aqueous solution to the mixed solution to promote the polymerization. Under heating at 85 °C for about 3–5 h, the mixed solution was solidified to form a mixture of well-distributed polyacrylates of Li, Ni, and Mn. Afterward the resulting polyacrylates were dried at 120 °C for 24 h. The obtained copolymeric precursor was then decomposed at 450 °C for 5 h in air to get the powders of a layered Li–Ni–Co–Mn–O. Then, the powders were finally calcined at 900 °C for 20 h in air to obtain homogeneous Li[Li0.2Co0.13Ni0.13Mn0.54]O2 nanocrystals.
:H2O = 70:30 wt%) under stirring, and then add a small amount of (NH4)2S2O8 aqueous solution to the mixed solution to promote the polymerization. Under heating at 85 °C for about 3–5 h, the mixed solution was solidified to form a mixture of well-distributed polyacrylates of Li, Ni, and Mn. Afterward the resulting polyacrylates were dried at 120 °C for 24 h. The obtained copolymeric precursor was then decomposed at 450 °C for 5 h in air to get the powders of a layered Li–Ni–Co–Mn–O. Then, the powders were finally calcined at 900 °C for 20 h in air to obtain homogeneous Li[Li0.2Co0.13Ni0.13Mn0.54]O2 nanocrystals.
Surface modification of the as-synthesized layered Li[Li0.2Co0.13Ni0.13Mn0.54]O2 with 3 wt% Al2O3 was carried out as follows: the as-prepared Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powder was firstly suspended in a solution containing the stoichiometric amount of Al(NO3)3·9H2O dissolved in distilled water. A solution of NH3·H2O was added dropwise to the suspension with stirring at 80 °C for 5 h. Thereafter, the powder was coated, filtered and dried at 100 °C overnight and then calcined at 300 °C for 4 h.
2.2. Structural characterization
To reveal the crystalline structure of the Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powders, X-ray diffraction (XRD) were carried out on a Shimadzu XRD-6000 diffractometer with Cu K radiation. The XRD spectra were collected in a range of 2θ values from 10 to 80 °C at a scanning rate of 2 deg min−1 and a step size of 0.02 °C. The morphologies of the as-synthesized samples were observed by scanning electron microscope (SEM, Sirion 2000, FEI) and the nanostructures of the samples were examined by transmission electron microscopy (TEM, JEM-2010FEF). The samples for TEM analysis were prepared by dispersing the sample powders in ethanol and releasing a few drops of the dispersed solution on a carbon film supported on a copper grid.2.3. Electrochemical measurements
Electrochemical measurements were carried out using CR2032 coin-type cells. The working electrodes were made by pressing a 0.8 cm2 thin film (containing 80 wt% Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powder, 12 wt% acetylene black, and 8 wt% polytetrafluoroethylene) onto a Al mesh. Electrochemical cells were assembled with the layered oxide as a cathode, a metallic lithium disk as an anode, a piece of Celgard 2400 porous film as separator. The electrolyte used in this work is a mixed solution of 1 M LiPF6 dissolved in a mixed solvent of ethylene carbonate (EC), dimethyl carbonate (DMC), and ethylene methyl carbonate (EMC) (1:1:1 by wt.). Charge-discharge experiments were performed galvanostatically with current density of 20, 100, 200 and 400 mA g−1 between 2.0 and 4.8 V on battery testers (Land CT2001A). The cells were cycled at room temperature to compare the cycling performances of the uncoated and coated samples. The cyclic voltammogram (CV) were measured by powder microelectrodes on an electrochemical station (CHI 660a) at a scanning rate of 0.1 mV s−1 with the voltage ranging from 2.0 to 4.8 V. And electrochemical impedance spectroscopy (EIS) measurements of all the samples were conducted at open-circuit voltage in the frequency range of 100 kHz to 0.05 Hz with AC voltage amplitude of 5 mV using an IM6 electrochemical impedance analyzer. Before testing EIS, the samples rested for 3 h at room temperature on cells charged to 4.8 V with 3 h rest during the 1st and 10th cycles.
3. Results and discussion
3.1. Synthetic reaction
The polymer-pyrolysis method used in this work is to synthesize the copolymeric precursor of the mixed metallic ions and then to pyrolyze the precursor into the required oxide structure. The synthetic chemistry for making Li[Li0.12Ni0.32Mn0.56]O2 has been well discussed in detail in our previous papers.28,29 In this work, we added the nitrates of Ni2+, Co2+ and Mn2+ into aqueous acrylic acid (AA) and used LiOH as a Li source and a neutralizer. After a solution polymerization, a copolymeric polyacrylate precursor is formed with Li+, Mn2+, Ni2+, and Co2+ ions distributed homogeneously in the polymeric skeleton (see the inset in Scheme 1), which can be directly pyrolyzed to produce nanocrystalline Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powders. This preparation process of the Li[Li0.2Mn0.54Ni0.13Co0.13]O2 powders is schematically illustrated in Scheme 1.![Schematic picture of the preparation process of the as-prepared Li[Li0.2Mn0.54Ni0.13Co0.13]O2 powders. Inset is EDS mapping of the copolymeric precursor.](/image/article/2012/RA/c2ra20122d/c2ra20122d-s1.gif) | ||
| Scheme 1 Schematic picture of the preparation process of the as-prepared Li[Li0.2Mn0.54Ni0.13Co0.13]O2 powders. Inset is EDS mapping of the copolymeric precursor. | ||
3.2. Structural features
The XRD patterns of the pristine Li[Li0.2Co0.13Ni0.13Mn0.54]O2 and the 3 wt% Al2O3-coated samples are shown in Fig. 1. All the strong diffraction lines can be indexed as a layered oxide lattice based on a hexagonal α-NaFeO2 structure with a space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The weak peaks between 20° and 25° are reflected by a monoclinic unit cell with a C2/m symmetry rather than a Rm lattice, due to a LiMn6 cation arrangement that occurs in the transition metal layers of Li2MnO3 regions. Therefore, the layered Li1 + xMO2 materials can be alternatively represented in a two-component “composite” notation as xLi2MnO3·(1 − x)LiMO2 (M = Ni, Co, Mn, etc).30 As can also be seen in Fig. 1, both the (006)/(102) and (108)/(110) peaks are well split, suggesting a well-defined layered structure formed in the lattice. In general, the integrated intensity ratio (R) of the (003) to (104) lines in the XRD patterns can be used to denote the degree of cation mixing in the Li-layers of these materials. If the R value is > 1.2, the cation mixing could be considered to be negligible.31 In the XRD pattern of the pristine sample, the relative intensity ratio of the (003) to (104) lines is about 1.62, suggesting that the disordered arrangement of Ni and Li ions in the Li-layers could be ignored. Compared with the pristine sample, the 3 wt% Al2O3-coated sample did not show any change in its XRD pattern without any reflections from the Al2O3-coated surface layer, which is possibly due to the very low content and poor crystallinity of the very thin Al2O3 layer coated on the surface of the material. This XRD feature indicates that the surface modification did not change the lattice structure of the bulk material.
m. The weak peaks between 20° and 25° are reflected by a monoclinic unit cell with a C2/m symmetry rather than a Rm lattice, due to a LiMn6 cation arrangement that occurs in the transition metal layers of Li2MnO3 regions. Therefore, the layered Li1 + xMO2 materials can be alternatively represented in a two-component “composite” notation as xLi2MnO3·(1 − x)LiMO2 (M = Ni, Co, Mn, etc).30 As can also be seen in Fig. 1, both the (006)/(102) and (108)/(110) peaks are well split, suggesting a well-defined layered structure formed in the lattice. In general, the integrated intensity ratio (R) of the (003) to (104) lines in the XRD patterns can be used to denote the degree of cation mixing in the Li-layers of these materials. If the R value is > 1.2, the cation mixing could be considered to be negligible.31 In the XRD pattern of the pristine sample, the relative intensity ratio of the (003) to (104) lines is about 1.62, suggesting that the disordered arrangement of Ni and Li ions in the Li-layers could be ignored. Compared with the pristine sample, the 3 wt% Al2O3-coated sample did not show any change in its XRD pattern without any reflections from the Al2O3-coated surface layer, which is possibly due to the very low content and poor crystallinity of the very thin Al2O3 layer coated on the surface of the material. This XRD feature indicates that the surface modification did not change the lattice structure of the bulk material.
![XRD patterns of the as-prepared pristine and 3 wt% Al2O3-coated Li[Li0.2Mn0.54Ni0.13Co0.13]O2 samples.](/image/article/2012/RA/c2ra20122d/c2ra20122d-f1.gif) | ||
| Fig. 1 XRD patterns of the as-prepared pristine and 3 wt% Al2O3-coated Li[Li0.2Mn0.54Ni0.13Co0.13]O2 samples. | ||
Scanning electron microscopy (SEM) photographs of Li[Li0.2Co0.13Ni0.13Mn0.54]O2 are shown in Fig. 2a and b. The SEM images reveal that the powders were comprised of homogeneous particles with diameters in a narrow range between 100–150 nm with only a few aggregations. To ensure the effective coating of Al2O3 on the surface of the layered oxide, high resolution TEM observation of the 3 wt% Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 was also carried out. As shown in Fig. 2c, the coated powders were composed of ultrafine particles with a uniformly distributed size of 100–150 nm, in accordance with the result of SEM. It can also be seen from Fig. 2d that the edge of the coated particle appears as a distinguishably translucent, porous and continuous coating layer with a thickness of 4 nm. The TEM image also clearly shows a good crystallinity of the bulk Li[Li0.2Co0.13Ni0.13Mn0.54]O2 material. Lattice fringes of the layer distance in the enlarged pictures (inset in Fig. 2d) are ∼0.47 nm apart, corresponding to the interplanar distance of the (003) planes with a rhombohedral structure, which agrees very well with the results obtained from XRD analysis. Undoubtedly, such nanosized particles must result in a high surface area and a short diffusion path for Li insertion/extraction and also for the diffusive transport of the oxygen ion vacancies, so as to enhance the electrochemical performance of this material, especially the high rate capability.
![(a, b) SEM of the as-prepared Li[Li0.2Mn0.54Ni0.13Co0.13]O2 particles. (c, d) TEM images of the 3 wt% Al2O3-coated Li[Li0.2Mn0.54Ni0.13Co0.13]O2 particles.](/image/article/2012/RA/c2ra20122d/c2ra20122d-f2.gif) | ||
| Fig. 2 (a, b) SEM of the as-prepared Li[Li0.2Mn0.54Ni0.13Co0.13]O2 particles. (c, d) TEM images of the 3 wt% Al2O3-coated Li[Li0.2Mn0.54Ni0.13Co0.13]O2 particles. | ||
3.3. Electrochemical performances
Fig. 3 compares the charge–discharge profiles of the pristine and surface-modified Li[Li0.2Co0.13Ni0.13Mn0.54]O2 in the first cycle at 20 mA g−1. Both the pristine and Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 electrodes show similar charge/discharge profiles. In the first charge, there is a clear change in the slope of the voltage/capacity profiles at ∼4.5 V, followed by a prolonged voltage plateau, which is characteristic of layered xLi2MnO3·(1 − x)LiMO2 compounds.32 It is now generally accepted that the charging reaction in the first sloping portion of the voltage curves corresponds to the oxidation of M component, i.e. Co3+ to Co3.6+ and Ni2+ to Ni4+ in this case, and the voltage plateau around 4.5 V is attributed to a loss of oxygen from the layered lattice as Li2O removal accompanying the generation of the oxide ion vacancies through cationic and anionic rearrangements at the end of the first charge.10 As can be seen in Fig. 3, the Al2O3-coated sample can realize a charge capacity of 324 mAh g−1 and a discharge capacity of 311.5 mAh g−1 in the first cycle at 20 mA g−1, giving a very high coulombic efficiency of 96.1%, whereas the pristine sample shows a much poorer coulombic efficiency of 82.7% with a lower discharge capacity of 290 mAh g−1 and a larger charge capacity of 351 mAh g−1. Obviously, such a surface modification can improve not only the initial charge–discharge efficiency but also the electrochemical utilization of the material. As far as we know, the coulombic efficiency of 96.1% for the Al2O3-coated sample obtained is the highest value reported so far.19,33–36 To account for the remarkably enhanced electrochemical performances, several causes have been proposed to reveal the effects of surface modification on the layered xLi2MnO3·(1 − x)LiMO2 electrodes. Firstly, the surface coating can effectively suppress the decomposition of electrolyte by isolating the oxidative high valence metal ions from the electrolyte solvent. On the other hand, a larger amount of oxide ion vacancies, which were left due to the loss of oxygen in the lattice during the first charge, can be retained in the oxygen sites with surface modification, as suggested by A. Manthiram et al.37 Such a strong retention of the oxide ion vacancies would provide sufficient active sites for lithium ion insertion during the discharge process.![Charge–discharge curves of the pristine and 3 wt.% Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples at 20 mA g−1.](/image/article/2012/RA/c2ra20122d/c2ra20122d-f3.gif) | ||
| Fig. 3 Charge–discharge curves of the pristine and 3 wt.% Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples at 20 mA g−1. | ||
The contribution of the oxide ion vacancies to the reversible capacity can also be quantitatively calculated from the initial charge and discharge capacities.36,37 The first charge capacity of the 3 wt% Al2O3-coated sample in the sloping portion (< 4.5 V) is 106 mAh g−1, corresponding to an extraction of 0.34 lithium ions, which is in good agreement with the stoichiometric oxidation of Co3+ to Co3.6+ and Ni2+ to Ni4+ as calculated from the chemical composition of the material. Thus, the charging reaction occurring in the sloping voltage region could be expressed as:
| Li[Li0.2Co0.13Ni0.13Mn0.54]O2 → Li0.66[Li0.2Co0.13Ni0.13Mn0.54]O2 + 0.34Li+ + 0.34e− | (1) |
Since the overall first charge capacity of the Al2O3-coated electrode is 324 mAh g−1, corresponding to an extraction of 1.03 lithium ions from the lattice, the charge capacity consumed in the voltage plateau region (> 4.5 V) must result in a further extraction of the remaining 0.67 lithium ions due to the loss of oxygen. Taking account of the irreversible capacity possibly caused by the decomposition of the electrolyte at high voltage, the maximum capacity contributed by the electrolyte decomposition is no more than 12.5 mAh g−1, because a very high reversible capacity of 311.5 mAh g−1(0.99 Li+), about 96.1% of its initial charge capacity, can be recovered at the subsequent discharge. From this capacity analysis, the actual amount of lithium ions extracted in the high voltage plateau should be 0.65 moles per unit of Li0.66[Li0.2Co0.13Ni0.13Mn0.54]O2 (0.99–0.34 = 0.65) due to the loss of oxygen. Thus, the initial charge reaction in the plateau region can be represented as:
| Li0.66[Li0.2Co0.13Ni0.13Mn0.54]O2 → Li0.01[Li0.2Co0.13Ni0.13Mn0.54]O1.675□0.325 + 0.65Li+ + 0.65e− + 0.16O2 | (2) |
Since the first irreversible capacity is very small, most of the oxide ion vacancies in the Al2O3-coated sample should be retained in the layered lattice at the end of first charge, which become active sites for subsequent lithium insertion/extraction reactions. At the first discharge, 0.34 Li+ ions were first re-inserted into the lattice along with electrochemical reduction of all the Ni4+ and Co3.6+ ions to Ni2+ and Co2+ ions. The remaining oxide ion vacancies in the lattice should allow additional 0.65 Li+ ions to insert into the active sites, which must lead to an electrochemical reduction of Mn4+ to Mn2.82+, due to the strict requirement for charge balance in the lattice. Therefore, the discharge reaction on the electrode can be expressed as:
| Li0.01[Li0.2Co0.13Ni0.13Mn0.54]O1.675□0.325 + 0.99Li+ + 0.99e− → Li[Li0.2Co0.13Ni0.13Mn0.54]O1.675□0.325 | (3) |
As discussed above, the high capacity and low initial capacity loss of the Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 electrode come from its strong retention of the oxide ion vacancies through a surface modification. This is possibly due to a densely and uniformly coated surface layer of Al2O3, as can be visualized from Fig. 2c and 2d, which can not only inhibit the decomposition of electrolyte, but also prevent the oxygen deficiency on the surface from reacting with the electrolyte to cause the loss of oxide ion vacancies and ionic rearrangement in the lattice. On the other hand, the nanosized particles prepared by our polymer-pyrolysis method may provide a short diffusion path of lithium and oxygen ions, leading to an enhanced utilization of the bulk material and thereby improving its reversible Li-storage capacity.
The surface effect of the coated oxide on the electrochemical behaviors can also be seen from the cyclic voltammetric (CV) responses. Fig. 4 shows the CV curves of the pristine and Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powders at a slow scan of 0.1 mV s−1 between 2.0 and 4.8 V. For the pristine sample, there were two oxidation peaks centered at ∼4.0 and ∼4.6 V in the first positive scan, corresponding to the oxidation of the M components (Ni2+, Co3+), and the loss of oxygen, respectively.32 In the reversal negative scan, there were three reduction peaks distinguishable at ∼4.3, ∼3.7 and ∼3.3 V for both the coated and uncoated samples. The reduction peaks at ∼4.3 and ∼3.7 V are apparently due to the reduction of Ni4+ and Co3.6+, while the peak at ∼3.3 V could be ascribed to the Mn4+ reduction due to the loss of oxygen in first charge. In the second scan, the CV features are significantly different from those observed in the first scan. The strongest peak at +4.6 V, suggesting the loss of oxygen in first scan, disappeared and a weak new peak emerged instead at ∼4.5 V. Because only the oxidation of Ni2+ and Co3+ can occur above 3.5 V in second cycle, the two oxidation peaks at 3.9 and 4.5 V must from the oxidation of Ni2+ and Co3+.10 The reversible redox of the Mn component can still be observed below 3.5 V in second scan. The CV features of the Al2O3-coated sample are similar to those of the pristine sample, except for a couple of new redox peaks appearing at 2.7–3.0 V. At this low potential region, the redox peaks most probably arose from the redox reactions of the Mn component, which is analogous to the charge–discharge reactions of LiMn2O4 at ∼2.8 V.38 In fact, a similar couple of peaks can be also observed, though somewhat vaguely, for the pristine sample at 2.7–3.2 V. Overall, this phenomenon implies that the Al2O3-coated sample exhibits better symmetric redox peaks than the pristine sample, suggesting a higher electrochemical reversibility of Li-insertion/extraction on the coated sample. This is possibly because that the Al2O3-coated surface layer is better than the SEI film formed during first charge for remaining the oxide ion vacancies and inhibiting the rearrangement of metal ions in the layered structure, which avoids the structural degradation of the crystal lattice of the material.
![Cyclic voltammograms of the pristine (a) and the Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 electrode (b) scanned at 0.1 mV s−1.](/image/article/2012/RA/c2ra20122d/c2ra20122d-f4.gif) | ||
| Fig. 4 Cyclic voltammograms of the pristine (a) and the Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 electrode (b) scanned at 0.1 mV s−1. | ||
The rate capability of the surface-modified Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples at a current density of 100, 200 and 400 mA g−1 are shown in Fig. 5. As it can be seen, the Al2O3-coated sample delivers a higher reversible capacity than the pristine sample at all the current rates. The Al2O3-coated sample exhibits a discharge capacity of 311.5, 275.6, 255.7 and 238.9 mAh g−1 while the pristine sample gives only 295, 254.5, 225.6 and 188 mAh g−1 at 20, 100, 200 and 400 mA g−1, respectively. The improved rate capability for the Al2O3-coated sample may result from the fact that the surface-coated layer may restrain the formation of undesired SEI film, leading to a higher Li transport rate in the surface region by reducing the charge transfer resistance. The Al2O3-coated sample also exhibits a higher cycling stability (83.8% capacity retention after 70 cycles at 20 mA g−1) than that of the pristine sample (74.6%) (Fig. 6). The slower capacity fading suggests a better retainability of the oxide ion vacancies not only in initial cycle but also in the subsequent cycles. In comparison with the well-documented data, the discharge capacities of the Al2O3-coated sample can reach 238.9 mAh g−1 at a high rate of 400 mA g−1, which is still higher than those obtained from the double-layer coated samples (∼230 mAh g−1 at 250 mA g−1).37 It can also be seen from Fig. 6 that although the pristine and Al2O3-coated samples demonstrate a large difference in their initial coulombic efficiencies (82.7% for pristine and 96.1% for the Al2O3-coated samples), both of them have a similar high coulombic efficiency of 98% since the second cycle and exhibit a high cycling stability in subsequent cycles (inset in Fig. 6). This excellent cyclability is possibly because these two materials, prepared by the polymer-pyrolysis, method have a high crystallinity without the cation mixing in their lattices.
![The charge–discharge curves of the pristine (a) and the 3 wt% Al2O3 coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples (b) at 20, 100, 200 and 400 mA g−1, respectively.](/image/article/2012/RA/c2ra20122d/c2ra20122d-f5.gif) | ||
| Fig. 5 The charge–discharge curves of the pristine (a) and the 3 wt% Al2O3 coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples (b) at 20, 100, 200 and 400 mA g−1, respectively. | ||
![Discharge capacities and coulombic efficiency of the pristine and 3 wt% Al2O3 coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples cycled at 20 mA g−1.](/image/article/2012/RA/c2ra20122d/c2ra20122d-f6.gif) | ||
| Fig. 6 Discharge capacities and coulombic efficiency of the pristine and 3 wt% Al2O3 coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 samples cycled at 20 mA g−1. | ||
To demonstrate the effect of Al2O3-coating on the electrochemical performance, EIS measurements of the pristine and Al2O3-coated samples were carried out as shown in Fig. 7. Based on the equivalent circuit given in Fig. 7, the value of the charge transfer resistance (Rct) and resistance for lithium ion diffusion in the surface film (Rf), including the solid-electrolyte interface (SEI) layer and surface-modification layer are obtained and listed in Table 1. It is easy to find that the values of both Rf and Rct decrease after surface modification with Al2O3, which reveals that the Al2O3-coated sample could suppress interaction between the cathode surface and the electrolyte and enhance in the kinetics of lithium-ion diffusion. Therefore, the Al2O3-coated sample can result in better capacity and rate capability by reducing the electrochemical resistance. In addition, during the 10 cycles, the Rf value of the pristine sample increased from 34.1 Ω (1st cycle) to 48.77 Ω (10th cycle) while the Rf value of the Al2O3-coated sample has hardly changed (from 23.45 Ω (1st cycle) to 27.73 Ω (10th cycle)), implying that the Al2O3-coated sample could form a more stable SEI film than the pristine one. Therefore, the surface modification with Al2O3 could also exhibit better cyclability.
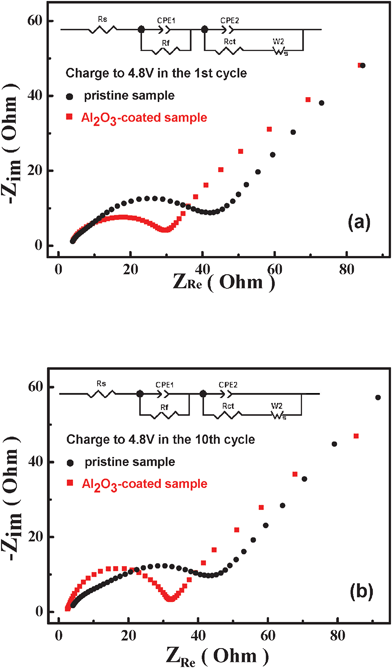 | ||
| Fig. 7 EIS plots of the pristine and Al2O3-coated sample. (a)1st cycle charge to 4.8 V; (b) 10th charge to 4.8 V | ||
| Al2O3-coated sample | Pristine sample | |||
|---|---|---|---|---|
| R f | R ct | R f | R ct | |
| 1st Charge | 23.45 | 193 | 34.1 | 1024 |
| 10th Charge | 27.73 | 59.7 | 48.77 | 324.5 |
Conclusions
In summary, we prepared high capacity Li[Li0.2Co0.13Ni0.13Mn0.54]O2 nanoparticles by a simple polymer-pyrolysis method. This layered material is proven to have a high crystallinity and a uniformly distributed nanosize of 100–150 nm by XRD and SEM/TEM characterizations. The charge–discharge experiments demonstrate that the as-prepared Li[Li0.2Co0.13Ni0.13Mn0.54]O2 powder has a high capacity (291 mAh g−1 at 20 mA g−1) and quite good cycling stability (74.6% capacity retention after 70 cycles at 20 mA g−1). After surface-coating with 3 wt% Al2O3, the coated sample gives a higher discharge capacity (311.5 mAh g−1) and initial coulombic efficiency (96.1%) and excellent cyclability with ∼83.8% capacity retention after 70 cycles. Such excellent electrochemical performances in the capacity, coulombic efficiency, and rate capability of the Al2O3-coated Li[Li0.2Co0.13Ni0.13Mn0.54]O2 electrodes result from: 1). well-distributed and crystallized nanoparticles without much cation mixing, which provide a short diffusion path and fast transport channels for lithium ion insertion/extraction reactions; 2). A strong retention of the oxide ion vacancies and a less metal ion rearrangement, leading to a higher reversible capacity; 3). An effective suppression of the irreversible decomposition of the electrolyte, improving the charge–discharge efficiency. In addition to the co-precipitation and sol–gel methods previously reported, the polymer-pyrolysis method along with the surface modification technique is simple and convenient for making high performance xLi2MnO3·(1 − x)LiMO2 materials, possibly for practical battery applications.Acknowledgements
We are grateful for the financial support provided by the National Basic Research Program of China (2009CB220100), the National high technology development program of China (863, No. 2011AA11A254) and the Fundamental Research Funds for the Central Universities.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. S. Kim, C. S. Johnson and M. M. Thackeray, Electrochem. Commun., 2002, 4, 205–209 CrossRef CAS.
- T. Ohzuku, M. Nagayama, K. Tsuji and K. Ariyoshi, J. Mater. Chem., 2011, 21, 10179–10188 RSC.
- J. Bareno, C. H. Lei, J. G. Wen, S. H. Kang, I. Petrov and D. P. Abraham, Adv. Mater., 2010, 22, 1122–1127 CrossRef CAS.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- Z. H. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novak, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS.
- B. Xu, C. R. Fell, M. F. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223–2233 CAS.
- A. Ito, D. C. Li, Y. Sato, M. Arao, M. Watanabe, M. Hatano, H. Horie and Y. Ohsawa, J. Power Sources, 2010, 195, 567–573 CrossRef CAS.
- J. Gao and A. Manthiram, J. Power Sources, 2009, 191, 644–647 CrossRef CAS.
- T. A. Arunkumar, E. Alvarez and A. Manthiram, J. Mater. Chem., 2008, 18, 190–198 RSC.
- S. T. Myung, K. Izumi, S. Komaba, Y. K. Sun, H. Yashiro and N. Kumagai, Chem. Mater., 2005, 17, 3695–3704 CrossRef CAS.
- B. C. Park, H. B. Kim, S. T. Myung, K. Amine, I. Belharouak, S. M. Lee and Y. K. Sun, J. Power Sources, 2008, 178, 826–831 CrossRef CAS.
- K. S. Tan, M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, J. Power Sources, 2005, 141, 129–142 CrossRef CAS.
- Y. Wu, A. V. Murugan and A. Manthiram, J. Electrochem. Soc., 2008, 155, A635–A641 CrossRef CAS.
- J. M. Zheng, J. Li, Z. R. Zhang, X. J. Guo and Y. Yang, Solid State Ionics, 2008, 179, 1794–1799 CrossRef CAS.
- H. Deng, I. Belharouak, C. S. Yoon, Y. K. Sun and K. Amine, J. Electrochem. Soc., 2010, 157, A1035–A1039 CrossRef CAS.
- J. Liu and A. Manthiram, J. Mater. Chem., 2010, 20, 3961–3967 RSC.
- J. Liu, Q. Y. Wang, B. Reeja-Jayan and A. Manthiram, Electrochem. Commun., 2010, 12, 750–753 CrossRef CAS.
- J. Liu, B. Reeja-Jayan and A. Manthiram, J. Phys. Chem. C, 2010, 114, 9528–9533 CAS.
- J. M. Zheng, X. B. Wu and Y. Yang, Electrochim. Acta, 2011, 56, 3071–3078 CrossRef CAS.
- M. G. Kim, M. Jo, Y. S. Hong and J. Cho, Chem. Commun., 2009, 218–220 RSC.
- D. H. Park, S. T. Lim, S. J. Hwang, C. S. Yoon, Y. K. Sun and J. H. Choy, Adv. Mater., 2005, 17, 2834 CrossRef CAS.
- L. H. Yu, H. X. Yang, X. P. Ai and Y. L. Cao, J. Phys. Chem. B, 2005, 109, 1148–1154 CrossRef CAS.
- L. H. Yu, Y. L. Cao, H. X. Yang and X. P. Ai, J. Solid State Electrochem., 2006, 10, 283–287 CrossRef CAS.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- K. M. Shaju, G. V. S. Rao and B. V. R. Chowdari, Electrochim. Acta, 2002, 48, 145–151 CrossRef CAS.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- C. S. Johnson, N. C. Li, C. Lefief and M. M. Thackeray, Electrochem. Commun., 2007, 9, 787–795 CrossRef CAS.
- C. S. Johnson, N. C. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef CAS.
- M. S. Park, J. W. Lee, Wonchang Choi, D. Im, S.-G. Doo and K.-S. Park, J. Mater. Chem., 2010, 20, 7208–7213 RSC.
- Y. Wu and A. Manthiram, Solid State Ionics, 2009, 180, 50–56 CrossRef CAS.
- Q. Y. Wang, J. Liu, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2009, 19, 4965–4972 RSC.
- S. Kuwabata, S. Masui and H. Yoneyama, Electrochim. Acta, 1999, 44, 4593–4600 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |