Exploiting the use of ionic liquids to access phosphorodiamidites†
Kerri
Crossey
a,
Christopher
Hardacre
*a,
Marie E.
Migaud
*b and
Sarah E.
Norman
a
aQUILL and School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, Northern Ireland BT9 5AG. E-mail: c.hardacre@qub.ac.uk; Fax: +442890874687
bSchool of Pharmacy, 97 Lisburn Road, Queen's University Belfast, Belfast BT9 7BL. E-mail: m.migaud@qub.ac.uk
First published on 25th January 2012
Abstract
A series of phosphorodiamidite reagents have been readily prepared using bis{(trifluoromethyl)sulfonyl}imide based ionic liquids and compared with their syntheses in conventional organic solvents. This method demonstrates a versatile procedure that allows access to both known and novel phosphorodiamidite reagents, whilst addressing issues such as moisture sensitivity and product selectivity present in current molecular based protocols. This method negates the need for reagent purification, whilst allowing for the reactions to be conducted at high concentrations.
Introduction
Phosphorodiamidites are a unique class of phosphorus based compounds containing a P–O and two P–N moieties. The presence of the P–N moieties in these molecules means that this emerging class of reagents have significant potential in a number of industrial and pharmaceutical processes. To date, phosphorodiamidites, notably 2-cyanoethyl-N,N,N',N'-tetraisopropylphosphoramidite, have played a key role in solid phase oligonucleotide synthesis.1 The use of these as synthetic precursors in oligonucleotide manufacture means that a number of nucleic acids are now at various stages of clinical trials for the treatment of a variety of disease states.2Beyond their use as synthetic precursors to oligonucleotides, phosphorodiamidites have been reported as starting materials for the synthesis of industrially relevant polymers as well as flame resistant materials such as adhesives, coatings and laminating materials.3 Phosphorodiamidites have also had some success as ligands in asymmetric catalysis;4 however, their phosphoramidite counterparts have been much more extensively explored in this area.5 In addition, there have been a limited number of examples concerning the use of phosphorodiamidites as nucleophilic starting materials to access phosphorus-carbon bond containing species6, including antiproliferative arylphosphonamidates.7
The limited use of phosphorodiamidites in industrial applications is, in part, due to their air and moisture sensitivity as well as the small number of analogues available. The conventional synthetic routes for these materials lead to purification issues due to unselective reactions and hydrolysis, most notably when incorporating diverse amino substituents.8 This has led to bis-diisopropylamino based phosphorodiamidites being the preferred reagents. These have increased stability and effective regioselectivity due to the steric hindrance of the amino groups. Whilst providing a route to access these relatively stable phosphorodiamidites, the use of the isopropylamino groups means that their chemical reactivity is reduced compared with phosphorodiamidites incorporating smaller amino substituents. For example, it has been shown that the presence of smaller amine groups at the phosphorus centre can result in increased coupling rates during the phosphitylation of nucleosides by chlorophosphoramidites.9 Similarly, Chamberlin has demonstrated that in automated RNA oligonucleotide synthesis, nucleoside phosphoramidites incorporating small amines were more readily oligomerised.10 However, due to the increased moisture sensitivity imposed by the nature of the substituents, access to the less hindered phosphorodiamidites is more challenging. As a result, these molecules are currently underused both in terms of basic research and applied technologies.
Current methods to access phosphorodiamidite derivatives require the treatment of excess PCl3 with a highly purified alcohol to yield the alkoxydichlorophosphine.11 Although a wide range of alkoxy groups have been investigated,12 cyanoethanol is the most commonly employed alcohol as deprotection of this group is readily accomplished via β-elimination with a mild base.13 The resulting highly reactive alkoxydichlorophosphine must then be distilled before it is reacted with an excess of a given amine, which also must be of the highest purity.11 Due to the commercial potential of these compounds, a more efficient process that also allows the incorporation of a wider range of amino substituents is highly desirable (Fig. 1). This will enable these compounds to have significantly wider applicability.
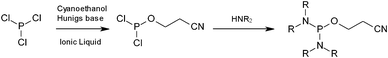 | ||
| Fig. 1 Synthetic pathway to phosphorodiamidite reagents. | ||
Ionic liquids (ILs) are a class of solvent which have gained significant interest over the last decade because of their unique properties such as a wide liquidous range and negligible vapour pressure.14 These properties mean that ILs are now used as viable alternatives to organic solvents in a number of industrial processes.15 Recently, ionic liquid media were shown to greatly enhance the hydrolytic stability of PCl316 as well as to enable excellent chemoselectivity when PCl3 is reacted with amines.17 Moreover, ILs have been shown to provide a unique medium for the synthesis, stabilisation and reactivity9 of a range of chlorophosphoramidites.18 Therein, only ILs based on the bis{(trifluoromethyl)sulfonyl}imide ([NTf2]−) anion were shown to provide an environment that enhanced both the hydrolytic stability and chemoselectivity of these halogenated phosphorus reagents. Ionic liquids with more coordinating anions such as triflate and mesylate reacted with the PCl3 whilst trifluorotris(perfluoroethyl)phosphate displayed poor solubility for PCl3 and, hence, was not suitable for this chemistry.
Within a given class of amine, phosphorodiamidites are thought to be more stable and less hydrolytically liable than their chlorophosphoramidite parents. This paper explores the potential of ILs as media for the synthesis, reaction and efficient storage of these compounds. Herein, we report the use of ionic liquids for the synthesis of a series of known and new phosphorodiamidites (Fig. 2), whereby enhanced yields and purities are achieved when compared with molecular solvents using the same reaction conditions.
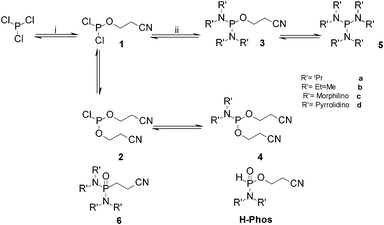 | ||
| Fig. 2 Phosphorodiamidite preparation: (i) Hünig's base (1eq), 3-hydroxypropionitrile (1eq), IL (2eq), 30 min, (ii) R'2NH (4eq), 120 min, N2. | ||
Results and discussion
The initial step in the synthesis of the phosphorodiamidites, 3a–d, involves the formation of alkoxydichlorophosphine, 1, which was carried out according to previously reported procedures.18 In agreement with those studies, 1 was obtained with yields of 91%, 89%, 89% and 75% in [C4dmim][NTf2], [C4mpyrr][NTf2], [C4py][NTf2] and [C4mim][NTf2], respectively. It should be noted that, unless otherwise stated, R = CH2CH2CN throughout the text. Other alkoxy groups, where R = CH3, CH2CH3, CH2CH2CH3, CH2(CH3)2, CH2C6H5, Ph, CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, have been investigated using this methodology; however, the yields and selectivities were reduced when compared with cyanoethanol. For example, in [C4dmim][NTf2], when R = CH2CH3, 1 was obtained in only 69% yield, with 20% attributed to the hydrolysis product, HP(O)(OR)Cl, compared with the 91% and no hydrolysis obtained when R = CH2CH2CN. This is attributed to the ability of the cyano group to complex water that may be present within the IL. This complexation means that the nucleophilicity of water is decreased, thus reducing the rate of hydrolysis of the hydrolytically unstable species present in the system.
CH2, have been investigated using this methodology; however, the yields and selectivities were reduced when compared with cyanoethanol. For example, in [C4dmim][NTf2], when R = CH2CH3, 1 was obtained in only 69% yield, with 20% attributed to the hydrolysis product, HP(O)(OR)Cl, compared with the 91% and no hydrolysis obtained when R = CH2CH2CN. This is attributed to the ability of the cyano group to complex water that may be present within the IL. This complexation means that the nucleophilicity of water is decreased, thus reducing the rate of hydrolysis of the hydrolytically unstable species present in the system.
Subsequent substitution with the nucleophilic amines was performed by adding the amine directly to the ionic liquid reaction mixture without purification of intermediate, 1. By comparison, the same procedure in molecular solvents requires careful purification of 1. In molecular solvents, careful work-up procedures are essential due to the hydrolytic instability of this compound and filtration of the ammonium chloride salt is required, as well as removal of the solvent.11 The generation of the ammonium chloride salt during reaction which is insoluble in the molecular solvents also means that highly dilute conditions are required. The fact that ammonium salts are much more soluble in the ILs means that only minimal ratio of ionic liquid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reactant is required for the reaction to occur, compared with the molecular solvent-based protocols.
:reactant is required for the reaction to occur, compared with the molecular solvent-based protocols.
Table 1 details the percentage distribution for the reaction mixtures after addition of a given amine in the absence of Hünig's base. Therein, the reactant amine is utilised as the base as well as the nucleophile. On replacing the amine with Hünig's base, there was no evidence for the formation of the desired phosphoramidite. For example, in the case of diisopropylamine, for a 1:2:2 ratio of 1:amine:Hünig's base, only mono substitution of 1 was observed with the main product being the chlorophosphoramidite P(OR)(NiPr2)Cl (181 ppm) obtained in 72% yield. This reduction in activity is thought to be due to the basicity of the secondary versus tertiary amine in the ILs. This is consistent with previously reported work on the synthesis of chlorophosphoramidites (P(OR)(NR'2)Cl).18 In all cases, using two equivalents of the amine gave higher yields of the desired chlorophosphoramidite product compared with the use of Hünig's base. In each case, the HCl formed following the nucleophilic substitution must be neutralised to ensure full conversion and prevent the reverse reaction. In the ionic liquid, secondary amines increase the degree of neutralisation compared with the Hünig's base thus resulting in a higher yield. As well as the formation of a range of P(OR)x(NR'2)3-x products, P(O)(R)(NR'2)2, 6, is also formed under certain conditions. This formation is attributed to the presence of the acidic β-hydrogens on the alkoxy group. These allow an internal rearrangement of the phosphorodiamidite to the corresponding alkylphosphonic diamide (Fig. 3).19
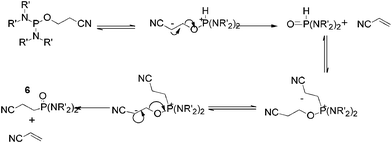 | ||
| Fig. 3 Proposed mechanism for the formation of 6. | ||
:4:0 1:amine:Hünig's base after 120 min. Two equivalents of ionic liquid were used in each reaction. Other refers to a range of decomposition products unless otherwise stated. The yields were determined from the 31P NMR peak integration ratio with the mole percentage corresponding to the ratio of the individual peak area to the total peak areas of all phosphorus containing species
| Solvent | Amine | Mole Percentage distribution (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| P(OR)(NR'2)23 | P(OR)2(NR'2) 4 | P(NR'2)35 | P(O)(R)(NR'2)26 | H-Phos | Other | |||
| a The remainder is attributed to 27% P(OR)(NiPR2)Cl (7) and 11% P(OR)3. | ||||||||
| [C4mim][NTf2] | a | Diisopropylamine | 50 | 22 | 2 | 20 | — | 6 |
| b | Ethylmethylamine | 67 | 11 | — | 6 | 4 | 12 | |
| c | Morpholine | 58 | 18 | 2 | 4 | 2 | 16 | |
| d | Pyrrolidine | 60 | 21 | — | 4 | 6 | 9 | |
| [C4dmim][NTf2] | a | Diisopropylamine | 58 | 21 | — | 9 | — | 12 |
| b | Ethylmethylamine | 57 | 17 | — | 13 | 7 | 6 | |
| c | Morpholine | 75 | 15 | 1 | 4 | 2 | 3 | |
| d | Pyrrolidine | 57 | 23 | — | 4 | 7 | 9 | |
| [C4mpyrr][NTf2] | a | Diisopropylamine | 64 | 16 | — | 15 | — | 5 |
| b | Ethylmethylamine | 62 | 17 | — | 7 | 4 | 10 | |
| c | Morpholine | 65 | 25 | 4 | 2 | — | 4 | |
| d | Pyrrolidine | 58 | 27 | — | — | 3 | 10 | |
| [C4py][NTf2] | a | Diisopropylamine | 48 | 15 | — | — | 11 | 21 |
| b | Ethylmethylamine | 64 | 19 | — | 5 | 4 | 8 | |
| c | Morpholine | 72 | 17 | 2 | 1 | 3 | 5 | |
| d | Pyrrolidine | 51 | 23 | — | — | 8 | 18 | |
| Dichloromethane | a | Diisopropylamine | 10 | 32 | — | — | 7 | 13a |
| b | Ethylmethylamine | 52 | 19 | 5 | — | 8 | 21 | |
| c | Morpholine | 44 | 17 | — | — | 10 | 24 | |
| d | Pyrrolidine | 32 | 16 | — | 12 | 30 | ||
| Diethyl ether | a | Diisopropylamine | 7 | 21 | — | — | 12 | 60 |
| b | Ethylmethylamine | 22 | 24 | — | — | 20 | 34 | |
| c | Morpholine | 7 | 15 | 11 | — | — | 67 | |
| d | Pyrrolidine | 18 | 40 | — | — | — | 42 | |
In contrast with the reported syntheses in molecular solvents, in the IL reactions, high concentrations of the reagents were used. Importantly, an additional mole equivalent of IL had to be added for every two equivalents of amine present in the reaction mixture. This was necessary to reduce the mass transfer issues due to the formation of two mole equivalents of ammonium chloride per mole of phosphorodiamidite formed. Although the ammonium chloride initially formed during the amination was soluble in the ionic liquid, as the reaction proceeds the viscosity of the reaction mixture increased to the point where it became significantly mass transfer limited when only 1 molar equivalent of ILs was used. Upon the addition of two molar equivalents of the IL at the beginning of the reaction, the salt remained in solution allowing the reaction mixture to remain fluid.
This synthetic method, i.e. using stoichiometric equivalents of 1 and amine with 0.04 M of solvent, was also examined using diethyl ether and dichloromethane (DCM). Both organic solvents had to be rigorously dried before use to prevent complete hydrolysis of 1 prior to the addition of the amine. Low yields of the desired phosphorodiamidites were achieved in diethyl ether, with 4a–4d being formed as the major products in all cases. When the reactions were performed in dry DCM, although the products 3a–3d were formed, the selectivity for the desired products was low for all amines studied. In addition, reactions in both organic solvents resulted in a marked increase in the number of decomposition products. For example, only 10% of 3a was observed in DCM with the remainder being 32% of 4a, 27% of P(OR)(NiPr2)Cl, 7, and 20% of decomposition products. This compares with only 5% decomposition and 64% selectivity to 3a in [C4mpyrr][NTf2]. In molecular solvents, the quality of the product can vary depending on the methodology implemented.20 For example, even after extensive optimisation, the best conditions to form 3a are reported to require a large molar excess of solvent and PCl3, where 3a is reported to be obtained in 48–70% yield;21 however, there was no indication of the purity of the product. Furthermore, for the morpholino derivative, 3c, Kierzek et al. reported 80% yield but only 60% purity which is compared with up to 90% purity in the present case (Table 2). In addition, the previously reported procedures for the synthesis of 3c required the use of extended reaction times, a large molar excess of PCl3 as well as high purity reagents.22 Using the methodology reported herein, no purification of the starting materials was required, the need for cooling the reaction vessel was also negated and all reactions were performed based on a one molar equivalent of PCl3. Furthermore, to the best of our knowledge, this is the first reported synthesis of the ethylmethyl and pyrrolidino derivatives, demonstrating the versatility of this procedure for the synthesis of both known and novel phosphordiamidite reagents.
| Ionic liquid | Phosphorodiamidite | |||
|---|---|---|---|---|
| 3a | 3b | 3c | 3d | |
| Yield/Purity | Yield/Purity | Yield/Purity | Yield/Purity | |
| [C4mim][NTf2] | 55/93 | 26/86 | 48/90 | |
| [C4dmim][NTf2] | 14/72 | 20/88 | 15/76 | |
| [C4mpyrr][NTf2] | 31/100 | 27/70 | 41/90 | 31/71 |
| [C4py][NTf2] | 43/81 | 17/84 | 10/69 | |
Isolation, stability and scalability
For 3b–3d, extraction with diethyl ether allowed the removal of the product from the ionic liquid. Distillation of these compounds from the IL proved unsuccessful, with the products decomposing even when using high vacuum. For 3a, both extraction with diethyl ether and distillation could be used, with the latter leading to the higher purity of 3a. The isolated yields and purities following solvent extraction and vacuum distillation are detailed in Table 2. All the phosphorodiamidites, 3a–d, were obtained in good purity which compares very favourably to the commercially available materials, as shown in the ESI† for 3a. Similarly, whilst the yields are only moderate, in all cases, they are significantly greater than those obtained in DCM, which are less than 20%. The best yields and purities were achieved for compounds 3a and 3c when synthesised in [C4mpyrr][NTf2], whilst [C4mim][NTf2] was the ionic liquid of choice for production of 3b and 3d.In order to investigate the scope of this procedure, a large scale reaction (0.1 mol based on PCl3, 80 cm3 [C4mpyrr][NTf2]) was performed to produce the morpholine derivative. The first step of the synthesis was complete within 60 min. The second step of the reaction required the slow addition of morpholine over a period of 30 min. After the amine addition, the reaction was stirred for a further 120 min at room temperature after which a 69% yield of 3c was achieved, which is consistent with the smaller scale reaction based on 2 mmol of PCl3. From this large scale reaction, extraction of 3c using anhydrous diethyl ether resulted in the phosphorodiamidite with a purity of 86% in 46% isolated yield.
Following the large scale reaction and extraction of the product, the ionic liquid was recycled via an aqueous wash of the IL/salt mixture. The IL was then dissolved in a minimal volume of DCM (50 cm3) and stirred with decolourising charcoal, filtered and concentrated before reuse. The DCM was necessary on the small scale to facilitate filtration of the ionic liquid; however, with larger volumes of IL DCM would not be needed due to the increased ease of separation of the solid and liquid.
On subsequent reactions, the products were obtained in similar yields and purities after the same reaction time as found for the original IL reaction. For example, on reacting 1 with morpholine, the [C4mpyrr][NTf2] ionic liquid could be recycled and reused 4 times before any appreciable decrease in yield and purity of 3c was observed. On the 5th reaction, the isolated yield reduced from 46% to 21% with a decrease in the purity of the product from 86% to 51%. This decrease in yield and purity may be attributed to the gradual loss of ionic liquid on recycle due to mechanical losses and adsorption on the charcoal or to the presence of residual ammonium chloride carried over during each recycle. Both scenarios have been shown, herein, to reduce the yield due to mass transfer issues. In addition, the presence of chloride salt increases the hydrophilicity of the ionic liquid and, therefore, its water content promoting the hydrolysis of the various phosphorus-containing reagents and intermediates.
Finally, current work within our laboratory has demonstrated that these phosphorodiamidite reagents can be used in combination with ball milling to successfully phosphitylate nucleosides and 2-deoxynucleosides in very high yields. This work will be reported in a future publication.
Conclusions
Herein, we have clearly demonstrated that ILs used in a 2:1 molar ratio with regards to PCl3 provide a suitable reaction media for the synthesis of highly moisture sensitive phosphorodiamidite reagents, without significant hydrolysis of the products, even when such compounds are known to be highly air and moisture sensitive. The use of ILs negates the need for intensive starting material purification, external cooling and large excess of molecular solvent, all of which are a prerequisite for the analogous reactions in molecular solvent.
Overall, ILs provide an easily scalable, facile and versatile approach to access a range of both known and novel phosphorodiamidite reagents in high purities. This methodology also significantly reduces the amount of waste solvent/PCl3 generated when compared with the reaction in conventional organic solvents. The phosphorodiamidites could be readily isolated from the reaction mixture via distillation in the case of the diisopropylamine derivative or via solvent extraction in the case of the morpholine, ethyl methyl and pyrrolidine derivatives.
Applicability of phosphorodiamidite reagents in wide industrial setting is a relatively underexplored area of phosphorus chemistry due to the limited availability of starting reagents. By easing the access to a variety of phosphorodiamidites, we have the scope for a wide variety of new methodologies to be realised.
Experimental
General procedures
The product distribution of the reactions for the synthesis of the phosphordiamidites was examined in situ by 31P NMR and 1H–31P coupled NMR. Four sets of parallel experiments were performed using 1-butyl-3-methylimidazolium bis{(trifluoromethyl)sulfonyl}imide ([C4mim][NTf2]), 1-butyl-2,3-dimethylimidazolium bis{(trifluoromethyl)sulfonyl}imide ([C4dmim][NTf2]), 1-butyl-1-methyl-pyrrolidinium bis{(trifluoromethyl)sulfonyl}imide ([C4mpyrr][NTf2]), 1-butylpyridinium bis{(trifluoromethyl)sulfonyl}imide ([C4py][NTf2]) as well as diethyl ether and dichloromethane. PCl3, diisopropylethylamine (Hünig's base) diisopropylamine, morpholine, ethylmethylamine, pyrrolidine, diethyl ether, dichloromethane and alcohols were all obtained from Aldrich and were used as supplied. The ionic liquids were prepared in house using standard literature methods from the appropriate halide salt.23 All ionic liquids were dried under high vacuum for 2 h prior to use. The water content was analysed using Karl Fischer titration. In each instance, the water content for the dried ILs were <0.04 wt%. The molecular solvents used were purified using standard literature procedures.24Spectroscopic details
All the 31P, 1H–31P, 1H and 13C nuclear magnetic resonance spectra were recorded on a Bruker Avance 300 MHz or 400 MHz NMR spectrometer at 25 °C. For all reactions an aliquot was transferred directly into the NMR tube with no addition of deuteriated solvents. The 31P NMR chemical shifts for all spectra were recorded in parts per million (ppm) relative to an external probe using a sealed capillary containing triethylphosphate (PO(OEt)3) in d6-DMSO (solvent used for locking/shimming optimization) inside the NMR tube. The PO(OEt)3 probe was referenced to 0.2 ppm.General experimental conditions
To a stirred solution of PCl3 in dried ionic liquid (2eq) under an inert atmosphere of N2, Hünig's Base (1eq) was added. After vigorous stirring for 5 min, alcohol (1eq) was added and the reaction stirred for a further 30 min. Nucleophilic amine (4eq) was then added and the reaction stirred for a further 120 min after which the reaction was generally complete. Extraction with 3 × 10 cm3 of anhydrous diethyl ether or short path distillation was then carried out to yield the desired phosphorodiamidite.Large scale synthesis of bis-(morpholino)-2-cyanoethoxyphosphite (3c) and recycle of the ionic liquid
To 0.2 mol (80 cm3) of [C4mpyrr][NTf2] was added 0.1 mol of PCl3 under N2. To this was added 0.1 mol of Hünig's base and the reaction was stirred for 5 min. 0.1 mol of cyanoethanol was then added and the reaction stirred for a further 30 min. 0.4 mol of morpholine was then added slowly via an addition funnel over 30 min. The reaction was then stirred for a further 120 min after which the named compound was extracted with 3 × 50 cm3 of anhydrous diethyl ether and concentrated to give a white solid in 46% isolated yield. The residual ionic liquid/salt mixture was subsequently washed with 100 cm3 of distilled H2O and the bottom layer separated. The bottom layer was then taken up in 50 cm3 of dichloromethane and stirred overnight with 5 g activated charcoal (Aldrich). The solution was filtered, concentrated and dried under high vacuum leading to the recovery of the [C4mpyrr][NTf2]. The ionic liquid was reused without further pretreatment and the reaction repeated.2-Cyanoethyl-N,N,N',N'-tetraisopropylphosphoramidite, 3a
Distilled from [C4mpyrr][NTf2] at 71 °C at 0.01 mmHg to give a colourless liquid (31%). 1H NMR (300 MHz, CDCl3) δ 1.17 (24H, t, J = 6.7 Hz, NCH(CH3)2), 2.61 (2H, t, J = 6.4 Hz, OCH2CH2CN), 3.49–3.58 (4H, NCH(CH3)2), 3.77 (dt, 2H, OCH2CH2CN, J = 7.3, 6.4 Hz). 13C NMR (75 MHz, CDCl3) 20.6 (d, J = 8.88 Hz), 23.8 (d, J = 5.77 Hz), 24.6 (d, J = 8.15 Hz), 44.6 (d, J = 12.4 Hz), 59.3 (d, J = 24.9 Hz), 118.0. 31P NMR (121 MHz, CDCl3) 123.7 (P(OR)(NiPr2)2). HRMS (ES, M + H+) calculated for C15H32N3OP 302.2361, found 302.2363.2-Cyanoethyl-N,N,N',N'-ethylmethylphosphoramidite, 3b
Extracted from [C4mim][NTf2] with diethyl ether to give a colourless liquid (55%). 1H NMR (300 MHz, CDCl3) 1.06 (6H, t, J = 7.1 Hz, 2 NCH2CH3), 2.56 (6H, d, 2 NCH3), 2.64 (2H, t, OCH2CH2CN), 3.12–2.86 (4H, m, 2 NCH2CH3), 3.78 (2H, dt, J = 7.6, 6.4 Hz, OCH2CH2CN). 13C NMR (75 MHz, CDCl3) δ 14.7 (d, J = 4.37 Hz), 20.6 (d, J = 7.46 Hz), 32.5 (d, J = 8.83 Hz), 44.4 (d, J = 28.3 Hz), 59.3 (d, J = 19.6 Hz), 118.2. 31P NMR (121 MHz, CDCl3) 135.0 (P(OR)(NEtMe)2). HRMS (ES, M + H+) calculated for C11H20N3O3P 218.1422, found 218.1428.Bis-(morpholino)-2-cyanoethoxyphosphite, 3c
Extracted from [C4mpyrr][NTf2] with diethyl ether to give a white solid (48%).1H NMR (300 MHz, CDCl3) δ 2.67 (2H, t, J = 6.17 Hz, OCH2CH2CN), 2.99–3.07 (2H, m, 2 NCH2), 3.60–3.63 (2H, m, 2 NCH2CH2O), 3.85–3.90 (2H, dt, J = 7.4, 6.2 Hz, OCH2CH2CN) . 13C NMR (75 MHz, CDCl3) δ 20.3 (d, J = 7.5 Hz), 45.1 (d, J = 14.9 Hz), 59.6 (d, J = 20.7 Hz), 67.9 (d, J = 6.3 Hz), 117.6. 31P NMR (121 MHz, CDCl3) δ 130.6. HRMS (ES, M + H+) calculated for C11H20N3O3P 274.1321, found 274.1310.Bis-(pyrrolidino)-2-cyanoethoxyphosphite, 3d
Extracted from [C4mim][NTf2] with diethyl ether to give a colourless liquid (37%).1H NMR (300 MHz, CDCl3) δ 1.75–1.79 (8H, m, CH2CH2NCH2CH2), 2.62 (2H, t, J = 6.2 Hz, OCH2CH2CN), 3.07–3.13 (8H, m, CH2CH2NCH2CH2), 3.81–3.88 (2H, m, OCH2CH2CN). 13C NMR (75 MHz, CDCl3) δ 20.9 (d, J = 6.6 Hz), 26.4 (d, J = 4.8 Hz), 47.0 (d, J = 15.8 Hz), 59.3 (d, J = 19.6 Hz), 118.2. 31P NMR (121 MHz, CDCl3) δ 133.6. HRMS (ES, M + H+) calculated for C11H20N3OP 242.1422, found 242.1412.References
- J. Michalski and W. Dabkowski, Top. Curr. Chem., 2004, 232, 93–144 CAS.
- (a) Y. S. Sanghvi, Z. Guo, H. M. Pfundheller and A. Converso, Org. Process Res. Dev., 2000, 4, 175–181 CrossRef CAS; (b) C. Xie, M. A. Staszak, J. T. Quatroche, C. D. Sturgil, V. V. Khau and M. J. Martinell, Org. Process Res. Dev., 2005, 9, 730–737 CrossRef CAS; (c) Manual of Antisense Methodology, ed. G. Hartmen and S. Endres, Kluwer Academic Publishers, Norwell, MA, 1999, p. 3 Search PubMed.
- (a) H. W. Coover and N. H. Shearer, US Patent, 1958, 2,856,390 Search PubMed; (b) H. W. Coover andN. H. Shearer, US Patent, 1957, 2,790,823 Search PubMed; (c) H. W. Coover and J. B. Dickey, US Patent, 1955, 2,275,371 Search PubMed.
- Y. Uozumi and M. Kimura, J. Org. Chem., 2007, 72, 707–714 CrossRef.
- B. L. Feringa and J. F. Teichert, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef.
- (a) P. I. Alimov and L. A. Antokhin, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1966, 1432–1434 Search PubMed; (b) T. K. Gazizov, L. K. Sal'keeva, E. K. Gafurov and A. V. Kazantsev, J. Gen. Chem. USSR. (Engl. Transl.), 1988, 1028–1029 Search PubMed; (c) T. K. Gazizov and L. K. Sal'keev, J. Gen. Chem. USSR. (Engl. Transl.), 1990, 1940–1941 Search PubMed; (d) V. S. Abramov and N.S. Il'ina, J. Gen. Chem. USSR. (Engl. Transl.), 1970, 96–98 Search PubMed.
- (a) L. K. Sal'keeva, M. T. Nurmaganbetova, E. V. Minaeva and B. Z. Kokzhalova, Russ. J. Gen. Chem., 2006, 76, 1397–1400 CrossRef CAS; (b) L. K. Sal'keeva, M. T. Nurmaganbetova, E. V. Minaeva and A. S. Kusainov, Russ. J. Gen. Chem., 2007, 77, 312 CrossRef CAS.
- N. Usman, K. K. Ogilvie, M. Y. Jiang and R. Cedergren, J. Am. Chem. Soc., 1987, 109, 7845–7854 CrossRef CAS.
- C. Hardacre, H. Huang, S. L. James, M. E. Migaud, S. E. Norman and W. R. Pitner, Chem. Commun., 2011, 47, 5846–5848 RSC.
- M. H. Lytte, P. B. Wright, N. D. Sinha, D. Nanda, J. D. Bain and R. A. Chamberlin, J. Org. Chem., 1991, 56, 4608–4615 CrossRef.
- (a) R. Gukathasan, M. Massoudipour, I. Gupta, A. Chowdhury, S. Pulst, S. Ratnam, Y. S. Sanghvi and S. A. Laneman, J. Organomet. Chem., 2005, 690, 2603–2607 CrossRef CAS; (b) P. R. J. Gaffney and C. B. Reese, J. Chem. Soc., Perkin Trans. 1, 2001, 192–205 RSC.
- (a) Y. Hayakawa, Bull. Chem. Soc. Jpn., 2001, 74, 1547–1565 CrossRef CAS; (b) L. Qian, Y. Xu, H. Arai, J. Aoki, T. M. Mc Intyre and G. D. Prestwich, Org. Lett., 2003, 5, 4685 CrossRef CAS.
- (a) Y. Tanaka, Y. Nakahara, H. Hojo and Y. Nakahara, Tetrahedron, 2003, 59, 4059–4067 CrossRef CAS; (b) D. Crich and V. Dudkin, J. Am. Chem. Soc., 2002, 124, 2263–2266 CrossRef CAS.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2007 Search PubMed.
- N. V. Plechkova and K. R Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- E. J. Amigues, C. Hardacre, G. Keane, M. E. Migaud and M. O'Neill, Chem. Commun., 2006, 72–74 RSC.
- E. J. Amigues, C. Hardacre, G. Keane and M. E. Migaud, Green Chem., 2008, 10, 660–669 RSC.
- E. J. Amigues, C. Hardacre, G. Keane, M. E. Migaud, S. E. Norman and W. R Pitner, Green Chem., 2009, 11, 1391–1396 RSC.
- J. Nielsen, J. E. Marugg, J. H. Van Boom, J. Honnens, M. Taagaard and O. Dahl, J. Chem. Research., 1986, 26–27 CAS.
- (a) J. Nielsen, J. E. Marugg, J. H. Van Boom, J. Honnens, M. Taagaard and O. Dahl, J. Chem. Research., 1986, 26–27 CAS; (b) D. Shamblee, W. Shming and W. Bing, US Patent, 2003, 60/388,224 Search PubMed; (c) S. E. Dinizo and J. M. Hardy, US Patent, 2003, 60/433788 Search PubMed; (d) Y. Ji, W. Bannwarth and B. Luu, Tetrahedron, 1990, 46, 487–502 CrossRef CAS; (e) Q. Gu and G. D. Prestwich, J. Org. Chem., 1996, 61, 8642–8647 CrossRef CAS.
- (a) Y. Ji, W. Bannwarth and B. Luu, Tetrahedron, 1990, 46, 487–502 CrossRef CAS; (b) Q. M. Gu and G. D. Prestwich, J. Org. Chem., 1996, 61, 8642–8647 CrossRef CAS.
- (a) R. J. Parry, M. R. Burns, S. Jiralerspong and L. Alemany, Tetrahedron, 1997, 53, 7077–7088 CrossRef CAS; (b) R. Kierzek and W. T. Markiewicz, Nucleosides Nucleotides, 1991, 10, 1257–1275 CrossRef CAS.
- P. Bonhôte, A. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- Purification of Laboratory Chemicals; ed. W. L. F. Armarego and C. L. L. Chai, Butterworth-Heinemann, 2003, 5th edition Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H NMR, 13C NMR and 31P NMR spectra following large scale synthesis in [C4mpyrr][NTf2] and isolation of bis-(morpholino)-2-cyanoethoxyphosphite (3c) and 1H NMR, 13C NMR and 31P NMR spectra of 2-cyanoethyl-N,N,N’,N’-ethylmethylphosphoramidite, 3b, and bis-(pyrrolidino)-2-cyanoethoxyphosphite, 3d, after solvent extraction. Also 31P NMR of 3a obtained from Aldrich. See DOI: 10.1039/c2ra20131c/ |
| This journal is © The Royal Society of Chemistry 2012 |