On the interaction of uranyl with functionalized fullerenes: a DFT investigation
Naresh K.
Jena
ab,
Mahesh
Sundararajan
a and
Swapan K.
Ghosh
*ab
aTheoretical Chemistry Section, Chemistry Group, Bhabha Atomic Research Centre, Mumbai, 400 085, India. E-mail: skghosh@barc.gov.in; Fax: +91 22 25505151; Tel: +91 22 25595092
bHomi Bhabha National Institute, Mumbai, 400 094, India
First published on 15th February 2012
Abstract
Understanding the interaction between uranyl and C60 can be useful for nuclear waste management processes. In this paper, we have investigated the binding of uranyl with bare C60 as well as functionalized C60 using density functional theory. It is observed that, the uranyl is only weakly bound to bare C60. On the contrary, functionalization of C60 with malonate ligand is shown to increase its binding ability to uranyl species. Further, we notice that pH could play an important role in the binding of uranyl at the malonate site. Our calculated geometries are in close agreement with the experimental data of the malonate uranyl complex. Further, based on the calculated binding energies, we show that a chelate type binding is more favorable in comparison to bidentate carboxylate binding.
Introduction
Due to the ever increasing demand for energy, nuclear energy is emerging as an indispensable source of alternate energy. Uranium, Neptunium and Plutonium are commonly encountered elements in the nuclear fuel cycle. The generation of nuclear power, however, comes with the penalty in the form of challenges associated with the disposal of radioactive nuclear wastes from the spent fuel. The radiation and toxicity generated from such nuclear waste contaminate the aquatic and geological systems which is of primary concern.However, there exist alternate ways of converting the toxic highly soluble UVI species to insoluble UIV species by means of electron transfer mechanisms. In fact, cytochrome c7 type bacteria present in D. Acetoxitans,1 iron-sulfur containing mineral surfaces (hematite)2 and humic acids3 derived from natural organic matter does convert UVI to UIV in an efficient manner via electron transfer processes.
Fullerene chemistry grew very rapidly during the last two decades due to the remarkable redox and energy storage properties associated with it.4–6 Furthermore, fullerene derivatives can be used as biologically active compounds in medicinal chemistry. Due to the poor solubility of C60, its application involving aqueous phase chemical processes has been hindered. Major efforts have, however, been invested to improve the solubility of C60 fullerene in aqueous phase. To this end, chemical functionalization bearing hydrophilic functional groups,7 encapsulation of C60 within γ-cyclodextrin8 and incorporation of C60 in micelles9 have improved the solubility in an aqueous environment. It is now being recognized that carbon nanotubes can also be a useful material for waste management processes.10–20
On the theoretical side, there has been a large increase in the number of computational investigations of actinide complexes, particularly those of uranium.22–31 The electronic structure and molecular geometry of the di-oxo cation of the three elements U, Np, and Pu have been studied at the Hartree Fock (HF),26 density functional theory (DFT)23,24,27,29 as well as correlated ab initio levels.22,28 In addition to the requirement of using high levels of theory and basis sets, modelling actinide ions is also quite challenging because of the need to account for solvation and relativisitic effects at some level. Fortunately, the DFT approach, with incorporation of relativistic effect via small core pseudo-potentials in conjunction with continuum solvation model seems to be sufficient and cost-effective. As far as accuracy of the results is concerned, calculations at this level are at least semi-quantitative.29
To this end, theoretical studies have also modelled these cations, especially uranyl, using various methods for the inclusion of aqueous solvation effects including variants of continuum models as well as by explicitly including the solvent molecules. The most studied structures are those which arise when the uranyl ion is hydrated, with the penta-aqua form being generally preferred to be the dominant species.25 The effect of bulk solvent is usually included via a continuum model, but there is an increasing interest in considering the actual nature of interaction with the nearest solvent molecules.29,30 Vazquez et al. modeled the uranyl acetate, carbonate and malonate complexes of uranyl in solution using DFT.31 We have recently investigated the interaction of uranyl with functionalized carbon nanotubes and found that the binding of uranyl is strong in the functionalized case than the unfunctionalized counterpart.32
Hence, there is need for further calculations in order to understand in more details the binding behavior of uranyl with nanostructures, which might help us in designing new nanomaterials that can be efficiently used for nuclear waste management. In this paper, we investigate the structures, vibrational frequencies and binding affinities of uranyl to malonate functionalized C60 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 ratio of functionalized C60 (malonate:uranyl) through DFT based calculations.
:1 and 1:2 ratio of functionalized C60 (malonate:uranyl) through DFT based calculations.
Computational details
All the geometries are optimized using BP86 functional in conjunction with def-TZVP basis for uranium, def2-TZVP basis for oxygen (of UO2) and def2-SV(P) basis set for the remaining atoms. For uranium, the core electrons are modeled with def-MWB pseudopotential. For energetics, we have used B3LYP functional with the same basis sets. Solvation effects are incorporated using COSMO continuum solvation model (using ε = 80) as implemented in TURBOMOLE.33 Such a cost effective strategy saves computational time and the predicted structures, vibrational spectra and redox properties are comparable to the experimental data.29Results and discussion
(i) Binding of uranyl at bare C60
We first present our results on binding of uranyl to bare C60 prior to considering functionalization. In Fig. 1, we have shown the five different binding sites of uranyl to C60. The first two binding sites are at the hexagonal carbon rings, where the uranyl can bind in two different orientations which is either parallel (μ2-||) or perpendicular (μ2-⊥) with respect to the C–C bond at the junction of two hexagons of C60 (Fig. 1). Similarly, we have made attempts to optimize the structures of the uranyl bound at the pentagonal carbon in both μ2-⊥ and μ2-|| modes with respect to the junction of the hexagon–pentagon bond of C60. However, the minimum energy structures are obtained only for the μ2-⊥ binding site, which will be discussed hereafter. In the fourth binding site, the uranyl ion can bind symmetrically to the pentagonal ring (μ5) of the carbon atoms. Finally, we have also confined the uranyl inside the C60 cage.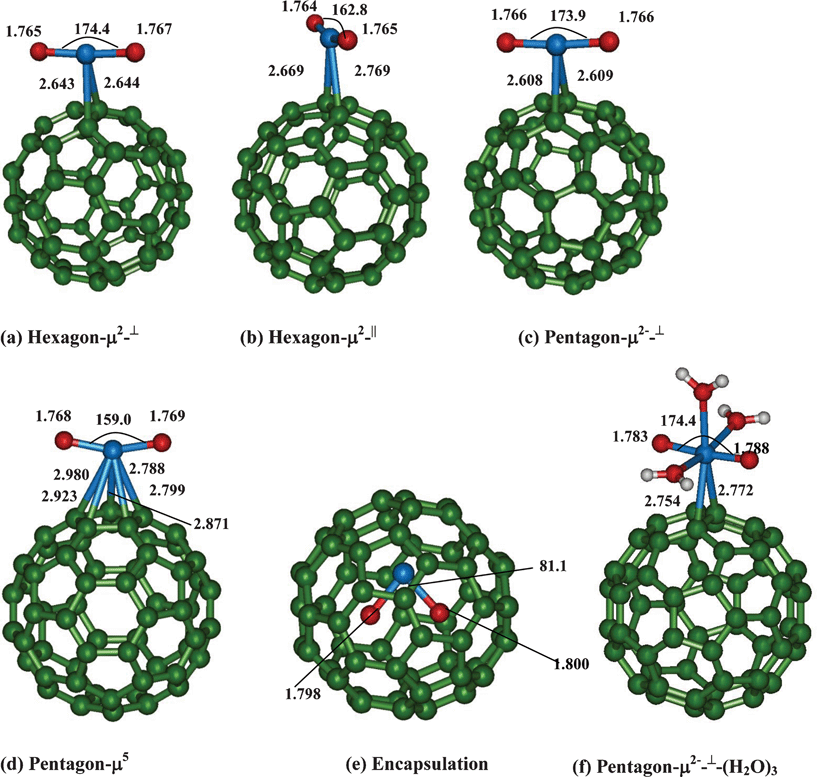 | ||
| Fig. 1 Binding of uranyl at various sites to bare C60 fullerene. | ||
For both hexagon and pentagon μ2⊥ binding, the calculated U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths (∼1.766 Å) and OUO (∼174.0°) bond angle are very similar. However, for the uranyl binding at the hexagon μ2|| binding mode, a strong bending of OUO (162.8°) is noted which is rather surprising. Within the three μ2 coordinations, the U–C bond lengths are somewhat shorter (by 0.06 Å) when uranyl binds at the pentagonal ring system (pentagon μ2⊥) as compared to the hexagons. Further, the two U–C bond lengths are symmetric for the μ2⊥ binding mode at both hexagons and pentagon ring systems.
O bond lengths (∼1.766 Å) and OUO (∼174.0°) bond angle are very similar. However, for the uranyl binding at the hexagon μ2|| binding mode, a strong bending of OUO (162.8°) is noted which is rather surprising. Within the three μ2 coordinations, the U–C bond lengths are somewhat shorter (by 0.06 Å) when uranyl binds at the pentagonal ring system (pentagon μ2⊥) as compared to the hexagons. Further, the two U–C bond lengths are symmetric for the μ2⊥ binding mode at both hexagons and pentagon ring systems.
For the μ5-binding, the uranyl bond lengths and bond angles are slightly elongated (∼0.002 Å) and more bent (by 4°) as compared to the three μ2-binding sites. The distances between U and C (of C60) for all the above four binding modes (excluding the encapsulation mode) have also been presented in Fig. 1. Between μ2 and μ5, the U–C bond strength for the latter species is weaker as indicative from their bond lengths. Attempts were even made to optimize the structure of the uranyl at the hexagonal ring system with μ6-binding. Unfortunately, we were unable to get the minimum energy species, due to the spontaneous formation of the μ2 coordination mode with the hexagons. Finally for the uranyl encapsulated species, the linear uranyl is much more elongated (1.798 Å) and very strongly bent (81.1°) as compared to other three binding sites.
All the four structures are characterized as minima and we find that the calculated vibrational spectra (symmetric (νsym) as well as asymmetric (νasym)) are slightly red shifted as compared to the penta-aqua–uranyl complex (Table 1). For instance, the calculated νsym and νasym of μ5 is about 20–25 cm−1 lower in energy as compared to the penta-aquo–uranyl complex. For the encapsulated uranyl adduct, this stretching is further reduced (854 cm−1 and 923 cm−1) due to UO bond elongation. For the μ2 binding motifs, the calculated vibrational spectra are only slightly downshifted as compared to the penta-aquo–uranyl complex.
| Binding mode | UO (Å) |
OUO (°) |
U–OMal (Å) | U–OH2O (Å) | UOνsym (cm−1) |
UOνasym (cm−1) |
|
|---|---|---|---|---|---|---|---|
| [UVIO2(C60–Mal–H2)(H2O)3]2+ | μ1-Carboxylate | 1.787 | 174.5 | 2.305 | 2.437–2.603 | 836.2 | 930.5 |
| [UVIO2(C60–Mal–H2)(H2O)3]2+ | μ2-Chelate | 1.788 | 173.9 | 2.404, 2.465 | 2.473–2.485 | 836.2 | 930.2 |
| [UVIO2(C60–Mal–H)(H2O)3]1+ | μ2-Carboxylate | 1.792 | 172.0 | 2.389, 2.391 | 2.482–2.497 | 831.4 | 926.2 |
| [UVIO2(C60–Mal–H)(H2O)3]1+ | μ2-Chelate | 1.795 | 172.1 | 2.197–2.470 | 2.497–2.549 | 825.4 | 918.7 |
| [UVIO2(C60–Mal)(H2O)3]0 | μ2-Carboxylate | 1.801 | 168.1 | 2.341 | 2.504–2.530 | 821.5 | 909.3 |
| [UVIO2(C60–Mal)(H2O)3]0 | μ2-Chelate | 1.809 | 167.7 | 2.222, 2.228 | 2.512–2.586 | 809.1 | 891.5 |
| [(UVIO2)2(C60–Mal)(H2O)3]2+ | (μ2)2-Carboxylate | 1.792, 1.788 | 173.4 | 2.400–2.408 | 2.479–2.486 | 829.6, 836.5 | 927.2, 929.2 |
Binding energies are calculated using the following equation,
[UO2]2+ + C60→ [UO2–C60]2+
Between the four species, pentagon-μ2-⊥ binding is the most stable species and the binding energy (COSMO-B3LYP) is found to be −6.27 kcal mol−1. Hexagon-μ2-|| is the least stable amongst the four binding patterns considered and the binding energy is estimated to be +4.93 kcal mol−1. It is also noted that hexagon-μ2-|| is less stable by 12.28 kcal mol−1 in comparison to the most preferred structure (pentagon-μ2-⊥). For the encapsulated adduct the binding energy is found to be 100.45 kcal mol−1 and it is less stable by 66.45 kcal mol−1 from the pentagon-μ2-⊥ structure. Further, the calculated binding is marginally favorable for hexagon-μ2-⊥ (−2.21 kcal mol−1) and unfavorable for pentagon-μ5 (+2.55 kcal mol−1). The weak binding of uranyl to C60 can be attributed to the inability of C60 to donate the charge to the lowest unoccupied molecular orbital (LUMO) of uranyl. As the charges of the carbons are very small (−0.001 to −0.005 au), the cation binding is very weak. Further, the binding efficacy of uranyl at various sites to bare C60 can be attributed to the extent of charge transfer from uranyl to C60. Indeed, it is known that in the case of metal atom-C60 interactions, charge transfer usually occurs from the metal centre to the C60 moiety, thus the metal atom attains partial ionic character.34 This concept has been successfully utilized in the field of hydrogen storage using metal decorated fullerenes. However, in our case, due to the di-positive charge of the uranyl species, further electron transfer from uranyl to C60 is highly unlikely. We find that the electron flow is from C60 fullerene to the uranyl species. Our Mülliken charge analysis does confirm the above statement. For instance, as compared to bare uranyl, the positive charge on the uranyl moiety for the most stable pentagon-μ2-⊥ species is smaller (1.28e−) as compared to least stable hexagon-μ2-|| species (1.36e−). The electron accepting nature of the C60 fullerene predominantly governs the fullerene chemistry, where the fullerene acts as a dienophile (electron acceptor) and forms favourable addition adducts with electron rich molecules like dienes.35 For these cases, the most preferable site of additions at the hexagonal junctions [6,6]. Conversely, in our case, we find that the [6,5] junctions is the preferable site of binding to uranyl. This signifies the basic differences between the interactions of C60 fullerene with different species.
As uranyl ion exists in aqueous phase with five water molecules, we have optimized a structure of uranyl with three water molecules bound to C60 (considering the most stable structure pentagon-μ2-⊥). The calculated uranyl bond lengths (1.783 Å) are lengthened due to the additional coordination of water molecules. However, the uranyl binding energies are unfavorable by more than 40 kcal mol−1, probably because the solvation energy of the uranyl ion is very high prior to C60 binding as compared to the adduct. We have also made attempts to optimize the structure of uranyl binding to C60 by keeping all five water molecules. Although the binding energies have reduced from 40 kcal mol−1 to ∼18 kcal mol−1, the relative trend is still the same. We note that the equatorial penta-coordination is only preferred over the hepta-coordination. The two additional water molecules are only interacting via hydrogen bonding with the coordinated water molecules (of the first coordination sphere).
Further, for effective separation, uranyl ion will be in aqueous phase and C60 will be in organic phase of the system. By attaching C60 with organic functional groups (such as malonate), one can increase the solubility of C60 and make it a better binder of uranyl in aqueous phase. From this part of the discussion it is quite clear that interaction of bare uranyl (along with explicitly hydrated species) with C60 is unfavorable. Hence, in the forthcoming sections, we focus on binding of uranyl with malonate functionalized C60.
(ii) Binding of uranyl at functionalized C60
Functionalization of C60 with carboxylic group such as malonic acid can improve the solubility and the strength of uranyl binding affinity to C60.21 We have functionalized C60 with a dicarboxylic acid, (malonic acid) and considered the uranyl binding to neutral, anionic and di-anionic form of malonate functionalized C60. All the three types of complexes which differ in the protonation states of carboxylic acid group are expected to be formed at different pH conditions. At low pH (pH = 1–3), both the carboxylate functional group will be protonated, and in semi-acidic pH (pH = 3–6), either one of the functional group is expected to be protonated (mono-anionic form), whereas at high pH (7–10), both the carboxylates will be deprotonated creating a di-anionic form.36We have considered uranyl binding to mono-dentate (neutral), bi-dentate (in both anionic forms) or chelate (all three forms) binding motifs of malonate functionalized C60 (Fig. 2, 3, 4).
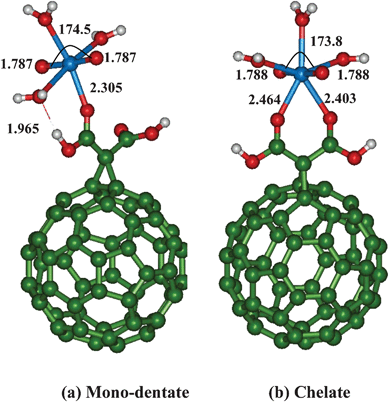 | ||
| Fig. 2 Binding of uranyl with neutral malonic acid functionalized C60 fullerene. | ||
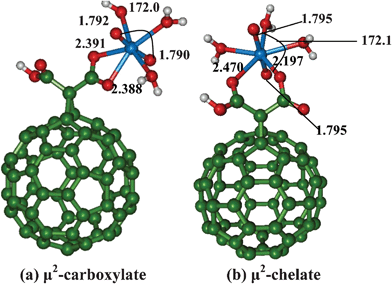 | ||
| Fig. 3 Binding of uranyl with mono-anionic malonate functionalized C60 fullerene. | ||
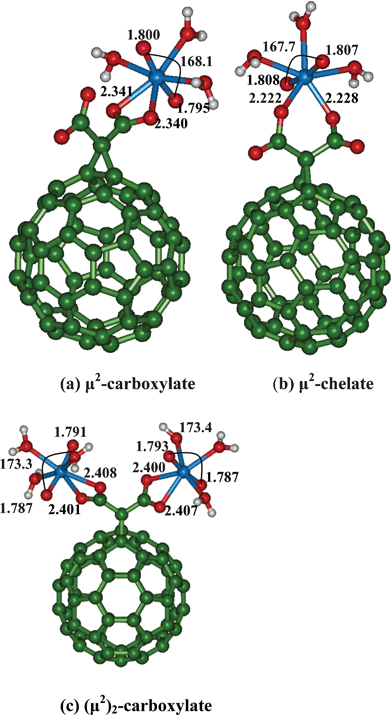 | ||
| Fig. 4 Binding of uranyl with di-anionic malonate functionalized C60 fullerene. | ||
For uranyl binding to neutral malonate functionalized C60 (Fig. 2), we find that the UO axial bond length is slightly longer for the chelate binding mode as compared to the mono-dentate binding mode. Similarly, the OUO bond angle is somewhat more bent (by 1°) in the chelate mode as compared to the same in mono-dentate binding. For mono-dentate carboxylate binding, during geometry optimization, we find that one water molecule is automatically detached from uranyl (2.751 Å) and now forms a hydrogen bonding with the carboxylate group of malonic acid if we consider tetra-aquo–uranyl with penta-coordination mode. Hence, we have considered only a tetra coordination site of uranyl (tri-aquo complex) with the mono-dentate carboxylate binding. This structure is characterized as minima (Fig. 2) and the geometrical parameters are given in Table 1.
For the mono-dentate carboxylate binding, the equatorially coordinated water molecule is hydrogen bonded (1.965 Å) to the carboxylate group, whereas in the case of chelate binding, no such weak interactions are noticed (Fig. 2). Further, asymmetric binding of two carboxylate oxygens to uranyl (2.404 Å and 2.465 Å) is noticed for chelating binding mode. Further, out of the monodentate and chelate modes, the latter uranyl binding mode is more favorable by ∼10 kcal mol−1.
For both mono-anionic and di-anionic species, we have considered two types coordination modes, one is of chelate type which we refer as μ2-chelate and the other is the symmetric bidentate carboxylate site denoted as μ2-carboxylate. The notation carboxylate or chelate μ2 arises from whether the bonded oxygen comes from the same carboxylic acid moiety or from two different carboxylic acids present in malonate.
As compared to the neutral malonic acid, the UO bond length are slightly weaker by 0.01 Å for both μ2 binding modes in case of corresponding mono-anionic form. Similarly, the OUO bond angle is also more bent by 2° in the latter binding sites. Between the two coordination modes, the μ2-carboxylate binding site is somewhat more stable by ∼3 kcal mol−1 as compared to the μ2-chelate binding site. Here again, we find the asymmetric U–O bond length (2.197 Å and 2.470 Å) is noted for chelate binding site due to the differing charges present on the oxygen atoms of the two carboxylates. Due to the strong coordination of malonate to uranyl, the coordinated water molecules become weakly bound (2.497–2.549 Å).
At high pH conditions, the coordination of uranyl will be at the completely deprotonated sites of malonate. Similar to the mono-anionic form, here we have two μ2-forms which are either bi-dentate μ2-carboxylate or μ2-chelate binding modes. Due to the double negative charge of the ligand, the binding of uranyl is expected to be stronger. Indeed, the UO bond length is more elongated (by 0.01 Å) as compared to the mono-anionic form and 0.02 Å with respect to neutral malonate form. Between the two binding sites, the μ2-chelate binding is stronger and energetically more favorable by ∼17 kcal mol−1 as compared to the μ2-carboxylate binding mode. The stronger binding of asymmetric μ2 mode to uranyl is also reflected in the OUO bond angle, which is more bent (167°) as compared to the μ2 carboxylate binding. Due to the strong coordination of negative charged ligand, we find that the water molecules are loosely bound (2.512 Å–2.586 Å).
If the concentration of uranyl is high, then the binding of uranyl to functionalized C60 in 1:2 (C60:uranyl) is possible. We have optimized this species (Fig. 4(c)), where the two uranyl ions bind via μ2-bidentate carboxylate functional groups of malonate. As compared to the μ2-carboxylate binding of malonate to one uranyl, we find that the binding of uranyl is somewhat weak which are reflected in the reduction of of UO bond length (1.792 Å) and the extent of bending of OUO bond angle being small (173.4°).
(iii) Vibrational frequencies
The favourable binding of functionalized C60 to uranyl will lead to elongation of UO bond as compared to penta-aqua–uranyl complex, and this weakening in the bond strength will be reflected in the vibrational frequencies corresponding to symmetric (νsym) and asymmetric (νasym) stretching of the UO bond. For all the complexes, the calculated UO νsym and νasym stretching modes (Table 1) are red-shifted as compared to the penta-aqua–uranyl complex. However, the extent of red-shift of the malonate–uranyl complex varies depending on the protonation state of the carboxylate group of the malonate.
Hence, at high pH (fully depronated), the uranyl bond lengths are elongated by more than 0.02 Å as compared to low pH (doubly protonated). Correspondingly, the uranyl stretching frequencies are downshifted by more than 25 cm−1. Between the two bidentate coordination modes, (carboxylate and chelate binding modes), both υsym and υasym μ2-binding are red shifted by nearly 20 cm−1. Hence, the vibrational frequencies of the uranyl bond can be used as a “fingerprint” to identify the type of coordination mode and the protonation state of malonate ligand can be understood at least semi-quantitatively.
(iv) Binding energies
The binding energy for the functionalized C60 to uranyl is evaluated using the following equation,[UO2(H2O)5]2+ + [Mal–Hn–C60]n−2 → [UO2–Mal–HnC60–(H2O)3]n + (H2O)2
We note that the formation of [UO2(H2O)3]2+ from [UO2(H2O)5]2+ is an endothermic process (36 kcal mol−1). The calculated binding energies (B3LYP/COSMO) for the neutral malonic acid to uranyl are unfavorable for both mono-dentate (19.1 kcal mol−1) and chelate (9.5 kcal mol−1) binding modes. However, for the case of mono-anionic and di-anionic malonate species (which are considered to be present in semi-neutral and high pH), the uranyl affinities are strongly favorable. For instance, for the mono-anionic malonate binding to uranyl, the calculated binding energies are −14.6 kcal mol−1 and −12.0 kcal mol−1 for μ2-carboxylate and chelate modes respectively. For the di-anionic species, the uranyl binding energies are strongly favorable for both μ2-carboxylate (−28.8 kcal mol−1) and chelate modes (−46.0 kcal mol−1). Similar to our earlier discussion, the possibility of binding of uranyl with all five water molecules and malonate is tested. We again find that two water molecules disassociated from uranyl and are weakly interacting with the coordinated water molecules. Further, the overall stabilization energy is not altered significantly. Hence, only penta-coordination mode of uranyl (with three water molecules) is favoured for both functionalized and bare C60.
Finally, in the case of 2:1 uranyl complex with deprotonated malonate, we find the binding energies to be somewhat weak (20 kcal mol−1 per uranyl) as compared to 1:1 ratio (28 kcal mol−1). Hence, we propose that favorable and efficient uranyl binding to malonate functionalized C60, is possible at high pH conditions.
We now compare our results with those of the recent experimental study using spectrophotometric measurements. Rao et al.37 carried out experiments (EXAFS) on complexation behavior of uranium(VI) with malonate at different temperatures and at differing pH (3.5 and 5.2). Our geometric predictions of uranyl bond lengths are in close agreement with those of the experimental data. Particularly, the calculated UO bond lengths from low pH (3.5) to semi-acidic pH (5.2), we find the elongation of 0.01 Å to be in close agreement with the experimental findings. In agreement with the experimental proposal, our calculations also suggest that chelate type binding with three equatorial water molecules is favored.
In the case of free malonate, more than one malonate molecule can interact with uranyl molecule. However, due to the presence of the C60 molecule, steric hindrance restricts the formation of such species which is again consistent with the experimental data.38
Conclusions
In this paper, we have considered the interactions between C60 and uranyl using DFT based calculations. The effect of pH is incorporated by carrying out calculations by varying the protonation states of malonate. Our calculations suggest that uranyl binding with bare C60 is not possible due to unfavorable binding energies. Upon functionalizing with malonate, the uranyl binding strength is improved upon increasing the pH of the medium. Further, we propose that chelate type binding is more favorable as compared to carboxylate binding. Our calculated geometries are also in close agreement with the experimental data. Although the formation of 2:1 and 3:1 ratio of malonate with uranyl are possible, it is highly unlikely to be formed in the case of malonate functionalized C60 due to steric hindrance. Functionalized fullerenes can be considered as an ideal building block for the design of materials and our study demonstrates that favourable binding of uranyl species with functionalized fullerenes may chart the road map for developing materials for effective nuclear waste management.
Acknowledgements
N.K.J gratefully acknowledges a senior research fellowship from Homi Bhabha National Institute, Department of Atomic Energy, India. The authors also acknowledge supercomputing facility of BARC for providing high performance parallel computing facility. S.K.G acknowledges DST-India for Sir J. C Bose fellowship and also support from the INDO-EU project MONAMI.References
- D. R. Lovley, E. J. P. Phillips, Y. A. Gorby and E. Landa, Nature, 1991, 350, 413–416 CrossRef CAS.
- G. Lefevre, S. Noinville and M. Fedoroff, J. Colloid Interface Sci., 2006, 296, 608–613 CrossRef CAS.
- P. M. Shanbhag and G. R. Chopping, J. Inorg. Nucl. Chem., 1981, 43, 3369–3372 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus and P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, New York, 1996, 870–917 Search PubMed.
- F. Giacalone and N. Martin, Chem. Rev., 2006, 106, 5136–5190 CrossRef CAS.
- S. A. Claridge, A. W. Castleman, Jr., S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 CrossRef CAS.
- I. Lamparth and A. Hirsch, J. Chem. Soc., Chem. Commun., 1994, 1727–1728 RSC.
- D. M. Guldi, H. Hungerbuehler and K.-D. Asmus, J. Phys. Chem., 1995, 99, 13487–13493 CrossRef CAS.
- Y. N. Yamakoshi, T. Yagami, K. Fukuhara, S. Sueyoshi and N. Miyata, J. Chem. Soc., Chem. Commun., 1994, 517–518 RSC.
- D. M. Guldi, J. Phys. Chem. A, 1997, 101, 3895–3900 CrossRef CAS.
- F. Belloni, C. Kutahyali, V. Rondinella, P. Carbol and T. Wiss, Environ. Sci. Technol., 2009, 43, 1250–1255 CrossRef CAS.
- J. Hu, C. Chen, X. Zhu and X. Wang, J. Hazard. Mater., 2009, 162, 1542–1550 CrossRef CAS.
- O. Moradi, K. Zare and M. Yari, Int. J. Nano Dim., 2011, 1, 203–220 CAS.
- J. P. Ruparelia, S. P. Duttagupta, A. K. Chatterjee and S. Mukherji, Desalination, 2008, 232, 145–156 CrossRef CAS.
- M. A. Atieh, O. Y. Bakather, B. S. Tawabini, A. A. Bukhari, M. Khaled, M. Alharthi, M. Fettouhi and F. A. Abuilaiwi, J. Nanomater., 2010, 2010, 1 CrossRef.
- O. Moradi, K. Zare, M. Monajjemi and H. Aghaie, Fullerenes, Nanotubes, Carbon Nanostruct., 2010, 18, 285–302 CrossRef CAS.
- N. Thamavaranukup, H. A. Hoppe, L. Ruiz-Gonzalez, P. M. F. J. Costa, J. Sloan, A. Kirkland and M. L. H. Green, Chem. Commun., 2004, 1686–1687 RSC.
- X. Wang, C. Chen, W. Hu, A. Ding, D. Xu and X. Zhou, Environ. Sci. Technol., 2005, 39, 2856–2860 CrossRef CAS.
- P. Wang, Patent Article. IPC8 Class: AG21F906FI, USPC, 2007, 588 18 Search PubMed.
- A. Schierz and H. Zanker, Environ. Pollut., 2009, 157, 1088–1094 CrossRef CAS.
- J. Soto-Guerrero and J. Havel, Polyhedron, 2003, 22, 1085–1090 CrossRef CAS.
- V. Vallet, U. Wahlgren, B. Schimmelpfennig, Z. Szabo and I. Grenthe, J. Am. Chem. Soc., 2001, 123, 11999–12008 CrossRef CAS.
- G. Schreckenbach, P. J. Hay and R. L. Martin, J. Comput. Chem., 1999, 20, 70–90 CrossRef CAS.
- N. Kaltsoyannis, Chem. Soc. Rev., 2003, 32, 9–16 RSC.
- M. Bühl, R. Diss and G. Wipff, J. Am. Chem. Soc., 2005, 127, 13506–13507 CrossRef.
- J. S. Craw, M. A. Vincent, I. H. Hillier and A. L. Wallwork, J. Phys. Chem., 1995, 99, 10181–10185 CrossRef CAS.
- G. A. Shamov and G. Schreckenbach, Acc. Chem. Res., 2010, 43, 19–29 CrossRef.
- D. Hagberg, G. Kalstrom, B. J. Roos and L. Gagliardi, J. Am. Chem. Soc., 2005, 127, 14250–14256 CrossRef CAS.
- J. A. Austin, M. Sundararajan, M. A. Vincent and I. H. Hillier, Dalton Trans., 2009, 5902–5909 RSC.
- P. J. Hay, R. L. Martin and G. Schreckenbach, J. Phys. Chem. A, 2000, 104, 6259–6270 CrossRef CAS.
- J. Vazquez, C. Bo, J. M. Poblet, J. de Pablo and J. Bruno, Inorg. Chem., 2003, 42, 6136–6141 CrossRef CAS.
- M. Sundararajan and S. K. Ghosh, J. Phys. Chem. A, 2011, 115, 6732–6737 CrossRef CAS.
- TURBOMOLE V6.0 2009, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com..
- K. R. S. Chandrakumar and S. K. Ghosh, Nano Lett., 2008, 8, 13 CrossRef CAS.
- P. Jayapal, M. Sundararajan, G. Rajaraman and P. Venuvanalingam, J. Phys. Org. Chem., 2008, 21, 146 CrossRef CAS.
- R. Colsenet, C. Gardiennet, B. Henry and P. Tekely, Angew. Chem., Int. Ed., 2002, 41, 4743–4745 CrossRef CAS.
- L. Rao, J. Jiang, P. Zanonato, P. Di Bernardo and A. Y. Garnov, Radiochim. Acta, 2002, 90, 581–588 CrossRef CAS.
- Y. Zhang, D. Collison, F. R. Livens, M. Helliwell, F. Heatley, A. K. Powell, S. Wocadlo and H. Eccles, Polyhedron, 2002, 21, 81–96 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |