New spinel oxide catalysts for visible-light-driven water oxidation†
Franziska
Conrad
a,
Matthias
Bauer
c,
Denis
Sheptyakov
d,
Stephen
Weyeneth
e,
Dominik
Jaeger
f,
Kathrin
Hametner
b,
Pierre-Emmanuel
Car
a,
Jörg
Patscheider
f,
Detlef
Günther
b and
Greta R.
Patzke
*a
aInstitute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland. E-mail: greta.patzke@aci.uzh.ch; Tel: +41 44 635 491; Fax: +41 44 635 6802
bLaboratory of Inorganic Chemistry, ETH Zurich, CH-8093, Zurich, Switzerland
cFachbereich Chemie, TU Kaiserslautern, D-67663, Kaiserslautern, Germany
dLaboratory for Neutron Scattering, ETH Zurich & PSI Villingen, CH-5232 Villingen PSI, Switzerland
ePhysik-Institut der Universität Zürich, CH-8057, Zurich, Switzerland
fLaboratory for Nanoscale Materials Science, EMPA Dübendorf, CH-8600, Dübendorf, Switzerland
First published on 30th January 2012
Abstract
Nanoscale Co–Mn–Ga spinels are promoted by the “synergistic” interaction of Co and Mn, thus paving the way to tailored and flexible catalyst design concepts for visible-light-driven water oxidation.
The quest for alternative energy sources and global climate control are major research challenges of the 21st century. Water splitting with visible-light-driven and robust heterogeneous catalysts would be a clean and direct chemical solution that attracts intense worldwide interest.1 We here present a new type of Co/Mn-WOCs that has been constructed along the lines of current research activities.
Mn-containing water oxidation catalysts (WOCs) are closest to nature's photosystem II,2 and several binary3 as well as ternary4 heterogeneous Mn-WOCs have been newly discovered. Recently, key relations between redox cycles of tetranuclear manganese WOC clusters and broader bio-geochemical phenomena have been established.5 Furthermore, a wide spectrum of cobalt compounds ranging from amorphous solids to molecular catalysts are active in water splitting.6 Cobalt phosphate-related WOCs with flexible self-repair features7 illustrate key criteria for artificial photosynthesis catalysts: robustness, abundance, stability and facile synthetic access. Structural analogies between PSII, molecular Co- and Mn-cubanes and cubic motifs in heterogeneous Co- and Mn-WOCs, such as Mn–Ca oxides,8 are being discovered.9 As this calls for further explorations, we have combined Co and Mn centers in a novel spinel-type WOC.
Nanostructured Co3O4 or Mn-oxide clusters in mesoporous matrices have shown excellent performance in water oxidation.3a Despite intense research efforts on individual Mn- and Co-based WOCs, little is still known about the catalytic activity resulting from their combination into a mixed metal structural motif. Heterogeneous cobalt catalysts often benefit from Mn-promoting effects,10 but this interesting phenomenon remains to be fully understood, and to be applied in WOC construction.
Spinel oxides of the MM′2O4 type are ideal targets for catalyst design due to their exceptional chemical stability and tuneability, which renders them flexible host matrices for catalytic centers. However, their full potential as visible-light-driven water splitting catalysts is not yet explored. Whilst Co/Mn-spinels are in the focus of current research,11e.g. for fuel cell materials or oxygen reduction/evolution electrocatalysts,12 Co–Mn–Ga spinels have never been investigated as such.13 Neither have gallium oxide spinels been used as visible-light-driven WOCs to date,14 despite recent progress on other Ga-oxide/oxynitride photocatalyst types.15
We thus introduce a new concept to link the rich catalytic chemistry of Co- and Mn-oxides into WOCs: both cations are embedded into a redox-inert and highly stable gallium oxide spinel matrix to promote their catalytic interaction. Here, we newly access nanoscale Co–Mn–Ga spinel WOCs with a rapid and facile microwave-hydrothermal (MW-HT) synthesis.16 MW-HT techniques excel through time- and energy-saving procedures which provide access to phases and nanoscale morphologies that are difficult to generate with conventional methods.17
Gallium nitrate, cobalt nitrate and/or manganese sulfate were dissolved in deionized water to yield nanoscale Co–Ga–, Mn–Ga– and Co–Mn–Ga-spinel materials, respectively, after 60 min of MW-HT treatment at 180 °C (Fig. 1). pH adjustment with NH3 or NaOH is the key control parameter to generate phase pure spinels: whereas Co–Ga and Co–Mn–Ga spinels require pH 10, Mn–Ga spinel emerges at a higher pH of 12 (Fig. S5 and S6, ESI†).
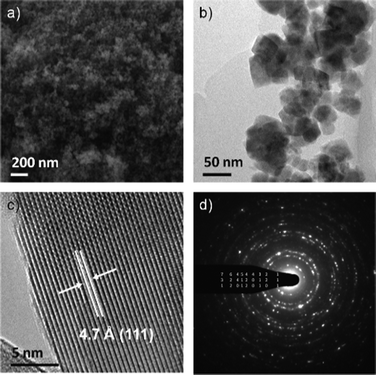 | ||
| Fig. 1 Representative SEM image of Co–Mn–Ga spinel (a), TEM image (b), HRTEM (c) and indexed ED (d). | ||
pH screens during the microwave-assisted formation of Co–Ga spinel (Fig. S5, ESI†) show that acidic media (pH 3) promote the formation of a different product type that is structurally related to GaOOH. Slightly higher pH values (4.3) induce an abrupt loss of crystallinity, and the spinel phase starts to emerge in neutral media (7.4). Optimum crystallinity of the Co–Ga spinel is finally achieved at pH 10. pH-dependent MW-HT synthesis of Mn–Ga spinel displays an analogous structural trend (Fig. S6, ESI†). PXRD patterns of the three pristine materials clearly indicate spinel formation (Fig. 2 and Fig. S5–7, S9–10, ESI†), and both particle size calculations and electron microscopy investigations cover the range between 15 and 40 nm (Fig. 1). This correlates with BET surface area values of 63 m2 g−1 (Co–Ga spinel), 39 m2 g−1 (Mn–Ga spinel) and 70 m2 g−1 (Co–Mn–Ga spinel).
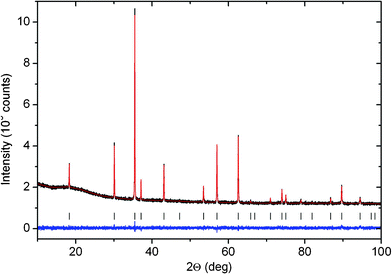 | ||
| Fig. 2 PXRD pattern of Co–Mn–Ga spinel after thermal treatment at 900 °C. | ||
All spinel-type nanomaterials were analyzed with ICP-MS as well as LA-ICP-MS and electron microprobe techniques, affording the composition Co0.58Mn0.95Ga1.47O4 for the nanoscale Co–Mn–Ga spinel. Electron microprobe and LA-ICP-MS analyses as well as STEM/EDX spot analyses (Fig. S12, ESI†) furthermore demonstrate that Co and Mn are homogeneously distributed within the spinel matrix.
Whilst the composition of Co–Ga spinel is close to CoGa2O4, Mn–Ga spinel is Mn-rich with a significantly higher Mn:Ga-ratio than hitherto known MnGa2O4 as the only well-defined spinel phase in the Mn/Ga/O-system. A separate structural study on MW-HT synthesized Mn–Ga spinel is thus in progress.
All three nanostructured spinels were compared with respect to their WOC performance under visible light irradiation. Catalytic tests were performed with [Ru(bpy)3]Cl2 as photosensitizer and S2O82− as sacrificial electron acceptor (Fig. 3, for details cf. experimental section).4 Oxygen evolution was observed for Co–Ga, Mn–Ga and Co–Mn–Ga spinels upon visible light illumination (λ = 470 nm) of the suspension with a LED lamp at very low power (4650 lux).
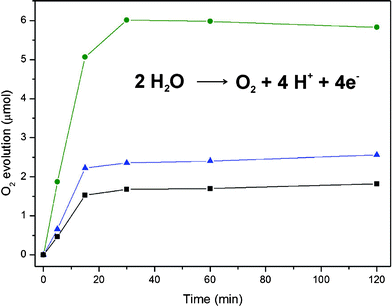 | ||
| Fig. 3 Visible-light-driven oxygen evolution for all three spinel-type catalysts (concentration 4.8 mmol L−1; ■ Mn–Ga, ▲ Co–Ga, • Co–Mn–Ga spinel). | ||
A slightly acidic Na2SiF6 buffered reaction medium (pH 5.8) prevents the accelerated degeneration of the Ru-photosensitizer at higher pH values. Variations of the reaction medium, e.g. acetate buffered solutions, did not afford satisfying results, and no photocatalytic activity was monitored in the presence of Fe3+ as the sacrificial electron acceptor.18 Maximum oxygen production rates are reached after 30 min and remain at this level afterwards. The optimal catalyst concentration for water oxidation was determined as 4.8 mmol L−1 for both Mn–Ga and Co–Mn–Ga spinel (cf. Fig. S15–17, ESI†). Even with a Co–Ga spinel concentration of 9.6 mmol L−1 at comparable BET surfaces of Co–Ga and Co–Mn–Ga catalysts, the maximum oxygen evolution of Co–Mn–Ga spinel is not reached.
A concentration-dependent catalytic test series (Fig. S15–17, ESI†) clearly demonstrate that Co–Mn–Ga WOC is superior to the binary spinel WOCs. Mn–Ga spinel displays the lowest WOC activity within the series. Although the Co–Ga spinel is more active than the Mn–Ga spinel, the higher transition metal content of Co–Mn–Ga WOC cannot be simply compensated by increasing the Co–Ga catalyst concentration (cf. Fig. S15–S17, ESI†). Most importantly, Fig. 3 illustrates that the oxygen evolution performance of Co–Mn–Ga WOC exceeds the sum of the binary spinel activities. These results point to a genuine “synergistic” interaction of Co and Mn centers within the spinel matrix.
Control experiments in the absence of catalysts did not lead to any oxygen evolution. All catalysts are stable under the reaction conditions and they could be recycled, which demonstrates the robust embedment of the interacting active centers within the spinel WOC matrix. Most importantly, the mixed Co–Mn–Ga spinel offers facile visible-light-driven operation with low power LEDs. This is another key advantage of the Co–Mn–Ga WOC over related heterogeneous Mn- and Co-catalysts working with high energy light sources (Ar ion lasers19 or Xe lamps4a,14).
To investigate the productive interaction of Co and Mn within the gallium oxide spinel matrix, a wide variety of analytical investigations was performed on the novel Co–Mn–Ga oxides. DTA/TG measurements show high thermal stability of Co–Mn–Ga spinel up to 1400 °C with negligible mass loss (Fig. S8, ESI†). All spinel samples were calcined (N2; Mn–Ga: 1000 °C, Co–Ga and Co–Mn–Ga: 900 °C) for detailed structural and magnetic investigations. The increased crystallinity of all three post-treated spinels goes hand in hand with microscale particle growth whilst phase changes were not observed (PXRD patterns in Fig. S7, S9 and 10, ESI†). Note that the catalytic activity of the thermally post-treated samples is lowered to ca. 1/4 compared to the pristine samples.
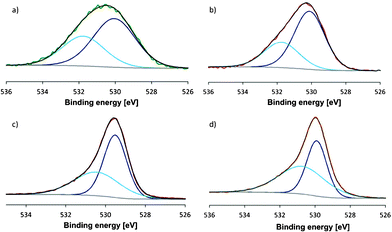
XPS (X-ray photoelectron spectroscopy) measurements on as-synthesized and calcined Co–Mn–Ga spinel did not exhibit significant differences and showed the presence of M(II) and M(III) species on the catalyst surface before and after thermal treatment (Fig. 4 and S13, Table S14). The mixed Mn valence is furthermore evident from the Mn 2p1/2 and 2p3/2 peak positions compared to manganese oxide references and from the multiplet splitting energy of the Mn 3s peak which falls within the characteristic range between Mn(II) and Mn(III).20
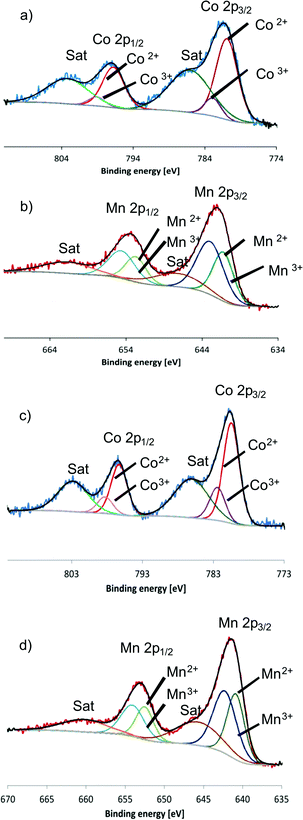 | ||
| Fig. 4 XPS spectra of pristine Co–Mn–Ga spinel: (a) Co 2p region and (b) Mn 2p region in comparison with XPS patterns of calcined Co–Mn–Ga spinel: (c) Co 2p region, (d) Mn 2p region. | ||
Likewise, the presence of both Co(II) and Co(III) states on the surface was assigned from detailed analyses of the Co 2p peaks (Fig. 4).
Local cationic coordination environments in all three post-treated bulk materials were investigated with X-ray absorption spectroscopy (XAS) measurements at the metal K-edges (Table 1, Fig. S2, ESI†). Oxygen coordination numbers obtained from EXAFS (extended X-ray absorption fine structure) allow the determination of tetrahedral and octahedral site occupancies based on their distinct M–O distances (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Co, Mn, Ga; cf.Table 1).21 Fractional differences from 100% arise from a fit procedure with minimal restrictions that is appropriate regardless of experimental errors (Table 1). Higher shells were fitted as well, but the obtained coordination numbers have to be treated with caution, because the similar backscattering properties of all three metals render them indistinguishable. Therefore, all spectra were only fitted with Ga–Ga contributions which leads to less reliable coordination numbers. Approx. 30% of the Ga atoms and 25% of the Co atoms are located on tetrahedral sites, whilst Mn is almost evenly distributed between tetrahedral and octahedral sites.
Co, Mn, Ga; cf.Table 1).21 Fractional differences from 100% arise from a fit procedure with minimal restrictions that is appropriate regardless of experimental errors (Table 1). Higher shells were fitted as well, but the obtained coordination numbers have to be treated with caution, because the similar backscattering properties of all three metals render them indistinguishable. Therefore, all spectra were only fitted with Ga–Ga contributions which leads to less reliable coordination numbers. Approx. 30% of the Ga atoms and 25% of the Co atoms are located on tetrahedral sites, whilst Mn is almost evenly distributed between tetrahedral and octahedral sites.
| Edge | Abs-Bsa | N(Bs)b | R(Abs-Bs)/Åc | Site occ. (x) | Oxid. state |
|---|---|---|---|---|---|
| a Abs = X-ray absorbing atom, Bs = backscattering atom (neighbour). b Number of backscattering neighbour atoms. c Distance between absorbing and backscattering atom. | |||||
| Ga K | Ga–O1 | 1.2 ± 0.1 | 1.90 ± 0.02 | IVGa: | |
| Ga–O2 | 5.2 ± 0.5 | 2.00 ± 0.02 | 0.30 ±0.05 | ||
| Ga–Ga1 | 7.2 ± 1.5 | 2.96 ± 0.03 | VIGa: | ||
| Ga–Ga2 | 5.2 ± 1.0 | 3.46 ± 0.04 | 0.86 ± 0.11 | ||
| Co K | Co–O1 | 1.0 ± 0.1 | 1.98 ± 0.02 | IVCo: | CoII: |
| Co–O2 | 3.4 ± 0.3 | 2.07 ± 0.02 | 0.25 ± 0.05 | 78% | |
| Co–Ga1 | 4.8 ± 0.5 | 2.93 ± 0.03 | VICo: | CoIII: | |
| Co–Ga2 | 7.6 ± 0.8 | 3.46 ± 0.04 | 0.56 ± 0.07 | 22% | |
| Mn K | Mn–O1 | 2.0 ± 0.4 | 1.95 ± 0.02 | IVMn: | MnII: |
| Mn–O2 | 3.3 ± 0.9 | 2.07 ± 0.02 | 0.50 ± 0.06 | 42% | |
| Mn–Ga1 | 5.3 ± 0.5 | 2.94 ± 0.03 | VIMn: | MnIII: | |
| Mn–Ga2 | 8.7 ± 0.8 | 3.47 ± 0.04 | 0.55 ± 0.07 | 58% | |
XANES (X-ray absorption near edge structure) spectra furthermore provide insight into the oxidation states of the metal cations through the use of defined references by a linear-combination XANES-fit (LCF).22 Given that MW-HT treatments promote oxidation of Co2+/Mn2+ containing precursors to M3+,23 Co- and Mn-oxides with various valence states were chosen as references. Only M(II)O and M(II, III)3O4 (MCo, Mn) references could be fitted to all three spinel materials and attempts to adjust M2O3 and MO2 were not successful. It is well known that the pre-edge peak shape is very sensitive to changes in the coordination, which might cause the deviations of the LC-XANES fits. Table 1 summarizes the results and slight divergences at the Mn K-edge which can be explained in terms of local structural differences between the reference oxides and the spinels under investigation. Co is mostly divalent (78% Co2+) whilst Mn2+ and Mn3+ are present in almost equal amounts. These results are in line with XPS investigations.
Both X-ray and neutron diffraction measurements done on the same Co0.58Mn0.95Ga1.47O4 spinel sample were analyzed by Rietveld refinement (Fig. 2 and Fig. S4). All three cation types were assumed to occupy both the 8a (1/8,1/8,1/8) and 16d (1/2,1/2,1/2) positions with soft constraints based on the initial chemical composition (S.G. Fd3m, a∼8.398 Å). This allowed a quantitative determination of the elements on the different sites (cf. Table S3, ESI†), assuming oxygen anions to be fully occupying the 32e (x,x,x) site with a refined value of x = 0.2599(1). In line with elemental analyses and electroneutrality requirements, the overall formula (Co2+0.46Mn2+0.54)[Co3+0.12Mn3+0.41(IVGa0.34VIGa1.13)]O4 was assigned. The M2+/M3+ ratios as well as the Ga site occupancies agree reasonably well with XANES and EXAFS data, respectively (Table S3). Site occupancies for M2+/M3+ cations with similar (back)scattering features could not be differentiated due to limited data resolution.
Finally, investigations of the magnetic susceptibility χm of Co0.58Mn0.95Ga1.47O4 for both zero-field cooled and field-cooled histories and the temperature dependence of χm provided interesting results (Fig. 5). The magnetic ordering below 25 K resembles ferrimagnetic order. From the slope of χm−1(T) above 100 K, well beyond the ferrimagnetic state, an effective magneton number of μeff = 4.25 μB was obtained by invoking the Curie–Weiss law.24 Due to four different magnetic ions in Co–Mn–Ga spinel (Mn2+/3+ and Co2+/3+) the overall magnetic character is a sophisticated interplay of 3d4, 3d5, 3d6, and 3d7 electrons. Importantly, the experimental result of 4.25 μB lies well within typical values of magnetic moments for these electron configurations.24 However, a theoretical calculation of the expected magneton number must involve both spin and angular momentum of each electronic level, i.e. a study in its own right.
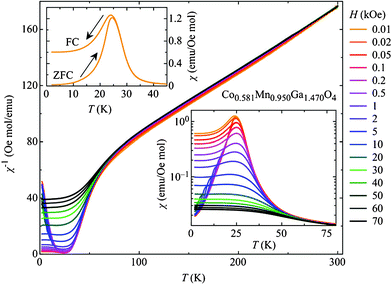 | ||
| Fig. 5 Temperature-dependent magnetic susceptibility χm measured in various magnetic fields for Co0.58Mn0.95Ga1.47O4. Main graph: inverse magnetic susceptibility; lower right: low temperature regime; upper left: different cooling procedures used. | ||
All in all, we introduce a new flexible, robust and visible-light-driven WOC type through synergism of Co and Mn, unifying the two most attractive elements for artificial photosynthesis to date. Understanding the structural and electronic details behind catalytic Co–Mn interactions, however, remains a general challenge,10,25 because Co–Mn spinels in their own right still require fundamental structural and synthetic investigations.11 Most interpretations of mixed Co/Mn-catalyst activities, e.g. in Fischer–Tropsch synthesis, CO oxidation or electrocatalytic oxygen evolution/reduction, are based on analyses of active surface oxygen species and on XPS monitoring of Co- and Mn surface oxidation states.10,12a The relative WOC activities found in the present study, namely Mn–Ga spinel < Co–Ga–spinel < Co–Mn–Ga spinel, correlate with the frequent use of Mn as a promoter for Co-based catalysts.10c
O 1s XPS spectra (Fig. 6) and BET investigations indicate that large surface area in combination with a high content of surface oxygen species are key performance parameters for the as-synthesized Co–Mn–Ga spinel. Generally, the O 1s peak can be deconvoluted into two peaks around 529 and 531 eV: peaks around 529 eV correspond to lattice oxygen species whilst peaks around 531 eV can be assigned to surface oxygen species.12a,26 XPS spectra of pristine Co–Mn–Ga WOC indicate the presence of higher amounts of surface oxygen species (Fig. 6a) than in the calcined sample (Fig. 6b). This enhanced surface reactivity in combination with a ten-fold higher surface area (70 m2 g−1vs. 6.8 m2 g−1) accounts for the superior WOC performance of as-synthesized Co–Mn–Ga spinel WOC. Furthermore, comparison of O 1s XPS spectra of pristine Co–Mn–Ga spinel with Co3O4 and Mn3O4 spinel references suggests that the mixed metal catalyst contains considerably more active surface oxygen species than the binary compounds (Fig. 6c, d). This is in line with the observed “synergistic” interaction of catalytic Co and Mn centers within the gallium spinel lattice.
 | ||
| Fig. 6 O 1s region of XPS patterns for: (a) as-synthesized Co–Mn–Ga spinel, (b) calcined Co–Mn–Ga spinel, (c) Mn3O4 and (d) Co3O4 references. | ||
New application-oriented tailoring perspectives for stable and tunable spinel-type WOCs are now opening up. Extensive screening of stable gallium spinel matrices loaded with Co and Mn centers in systematically varied ratios and oxidation states will provide deeper insight into their interplay. Follow-up studies on the WOC properties of Co–Mn spinels are now in progress in order to evaluate the role of trivalent and redox-inactive matrix cations on the catalytic interaction of transition metal centers.
In summary, we paved the way to a novel type of low-cost, environmentally friendly and abundant class of robust Co/Mn-oxide catalysts for water oxidation.
Experimental section
Catalyst preparation
Gallium nitrate (Ga(NO3)3·xH2O, Aldrich, 99.9%) was mixed with manganese sulfate (MnSO4·4H2O, Analar, > 97%) and cobalt nitrate (Co(NO3)2·6H2O, Fluka, > 98%) without further purification and the metal ratio (Ga:X) was adjusted to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Synthesis of mixed Co–Mn–Ga spinel started from a metal ratio of Co:Mn:Ga = 1:1:4. Precursor materials were dissolved in 15 mL deionized water using 95 mL CEM Omni Teflon vessels under thorough stirring, and the pH was adjusted with aqueous ammonia (25% NH3, Merck, GR for analysis) or freshly prepared 4 M NaOH solution (NaOH pellets, Acros Organics, > 97%) to the requested values. Reactions were carried out in a MARS5 microwave (CEM cooperation) with five vessels per synthesis run. A ramp to temperature mode with different ramp times (referred to as r) was selected for heating, followed by a constant temperature period (abbreviated as holding time h) and finalized by a 30 min cool down period (denoted as c). Pressure and temperature control were maintained with a reference vessel equipped with appropriate sensors, and additional temperature control of all vessels was done with IR sensors, whilst overheating was prevented by constantly stirring the reaction mixture and automatic power adjustment to stabilize the temperature in a ± 5 °C interval. A typical synthesis was conducted as follows: r = 30 min, h = 60 min (180 °C), c = 30 min. The obtained products were washed with deionized water, centrifuged several times for 10 min each (5000 rpm, Eppendorf 5804) and air dried for 2 h at 80 °C.
:1. Synthesis of mixed Co–Mn–Ga spinel started from a metal ratio of Co:Mn:Ga = 1:1:4. Precursor materials were dissolved in 15 mL deionized water using 95 mL CEM Omni Teflon vessels under thorough stirring, and the pH was adjusted with aqueous ammonia (25% NH3, Merck, GR for analysis) or freshly prepared 4 M NaOH solution (NaOH pellets, Acros Organics, > 97%) to the requested values. Reactions were carried out in a MARS5 microwave (CEM cooperation) with five vessels per synthesis run. A ramp to temperature mode with different ramp times (referred to as r) was selected for heating, followed by a constant temperature period (abbreviated as holding time h) and finalized by a 30 min cool down period (denoted as c). Pressure and temperature control were maintained with a reference vessel equipped with appropriate sensors, and additional temperature control of all vessels was done with IR sensors, whilst overheating was prevented by constantly stirring the reaction mixture and automatic power adjustment to stabilize the temperature in a ± 5 °C interval. A typical synthesis was conducted as follows: r = 30 min, h = 60 min (180 °C), c = 30 min. The obtained products were washed with deionized water, centrifuged several times for 10 min each (5000 rpm, Eppendorf 5804) and air dried for 2 h at 80 °C.
To improve their crystallinity, the gallium spinel samples were calcined in a tube furnace (Nabertherm RHTH 40-600/16) in nitrogen for 4 h. All samples were thoroughly grinded and placed in an alumina crucible which was transferred into the tube, flushed with nitrogen for 2 h and slowly heated to the desired temperature (100 °C h−1).
Catalyst characterization
X-Ray powder diffraction patterns (XRD) were recorded on a STOE STADI P diffractometer (transmission mode, Ge monochromator) with Cu Kα1 radiation. The neutron diffraction experiment was carried out with the high resolution powder diffractometer HRPT at the spallation neutron source SINQ (PSI-Villigen, Switzerland) with the neutron wavelength λ = 1.1545 Å at room temperature. The morphology of the substances was examined with scanning electron microscopy (SEM) performed on a LEO 1530 (FEG) microscope. Samples were dispersed in ethanol and applied on a silicon wafer. For transmission electron microscopy (TEM), the material was deposited on a perforated carbon foil supported on a molybdenum grid and investigated on a FEI CM30ST (LaB6 cathode). In the STEM mode, the electron beam was placed on selected areas, and an elemental analysis with energy-dispersive X-ray spectroscopy (EDXS, EDAX detector) was performed there. Specific surface areas of the samples were investigated with an adsorption isotherm of nitrogen at 77 K based on the Brunauer–Emmett–Teller method (BET; Quadrasorb SI, Quantachrome). Samples were degassed for 4 h at 150 °C prior to BET measurements. Determination of the metal ratios was performed with LA-ICP-MS methods. Analyses of the pressed powder samples were carried out using an 193 nm ArF excimer laser ablation system (Lambda Physik, Göttingen, Germany) coupled to an ICP-MS (DRC II +, Perkin Elmer, Norwalk, USA; for details cf. Table S1). Further elemental analysis was performed with a pressure digestion method followed by ICP-OES. 10 mg of the sample was dissolved in 3 mL HNO3 (suboiled) and 100 μL HCl (Merck suprapure) and heated within 20 min to 100 °C, followed by heating to 200 °C within 40 min and holding at this temperature for 30 min. The solution was afterwards cooled to 20 °C within 80 min and diluted with millipore water to a volume of 100 g. Samples were analyzed with an ICP-OES spectrometer (SPECTRO ARCOS, SPECTRO Analytical Instruments GmbH, Kleve Germany) with the following device details: 1400 W, 12 L min−1 Ar cooling gas, 1 L min−1 Ar help gas, 0.84 L min−1 atomizing gas and a crossflow atomizer. The measured element specific lines in nm were (repetition of each line in triplicate): Co (230.786, 237.862, 238.892, and 282.616), Ga (141.44 and 417.206) and Mn (257.611, 259.373, 260.569 and 294. 921). Electron microprobe analyses were performed on a JEOL JXA-8200 instrument, equipped with a SE-and a BSE-detector, 5 WDS crystal spectrometer and an EDS-analyser.TG/DSC investigations were carried out with a TG 449 Jupiter (Netzsch) in nitrogen atmosphere using different heating rates (5, 10 and 20 K min−1). Dynamic heating was performed to 400 °C, followed by a constant temperature mode for 2 h (400 °C) to completely remove any surface water. Afterwards, samples were dynamically heated to 1400 °C and kept at this temperature for 2 h. Sample powders for XPS measurements were suspended in ethanol and then dried on a glass plate so that dense coverage areas of the particles could be investigated. The Quantum 2000 Scanning XPS Microprobe spectrometer from Physical Electronics was operated with a monochromatic Al Kα source. Both the grainy morphology of the samples as well as the risk of sputter damaging precluded the use of sputter cleaning. Measurements were taken with a 100 μm probe size and a power of 25 W. The pass energy was set to 58.7 eV. To minimize sample charging effects during the measurements, an electron flood gun operated at 2.5 eV and an ion neutralizer using Ar+ of ca. 1 eV were used. These precautions minimize the fluctuations of the binding energy values due to charging to less than 1 eV. To allow for unambiguous comparison, the spectra within one set for a given sample are aligned to C 1s being set at 284.8 eV, as recommended by international standards.27 Spectra were evaluated using Physical Electronics' software package Multipak (v. 8.2). The spectra of samples with a finite density of states at the valence band are fitted with asymmetric peak shape, while those showing insulating behavior are approximated by Gauss-Lorentz functions. Magnetic susceptibilityχm was recorded on a Quantum Design SQUID magnetometer MPMS-XL. Measurements were performed for both zero-field cooled (ZFC) and field-cooled (FC) histories.
X-Ray absorption measurements were performed at the XAS beamline at the Ångströmquelle Karlsruhe (ANKA). The synchrotron beam current was between 80–140 mA at 2.5 GeV storage ring energy. A Si(111) double crystal monochromator was used for measurements at the manganese (6.540 keV), cobalt (7.709 keV) and gallium (10.367 keV) K-edge. The second monochromator crystal was tilted for optimal harmonic rejection. Samples were pressed into pellets using cellulose as stabilizer. The amount of sample used was adjusted to an edge jump between 0.3–0.8 (for details on data evaluation cf. ESI†).
Catalytic tests
Photocatalytic O2 evolution was performed using a standard [Ru(bpy)3]2+/S2O82− protocol. A stirred catalyst suspension (different concentrations, see Fig. S15–17) with 4.85 mg [Ru(bpy)3]Cl2, 7.7 mg Na2S2O8 in 6 mL buffered Na2SiF6 solution (20 mmol L−1, Na2SiF6, pH 5.8) was purged with He for 40 min to remove oxygen in a 10 mL headspace vial (with alumina crimp cap and rubber septum, BGB Analytik, Switzerland). The light source was a 470 nm high-flux LED from Rhopoint Components Ltd. (OTLH-0010-BU) with a CPC reflector for Shark LED, usually operated at usually 4650 lux using a TES 1332A digital lux meter. In regular intervals 100 μL of the reaction headspace were sampled with a gas tight microlitre syringe (Hamilton 1825 RN) using a gas chromatograph (Varian CP-3800) with He as carrier gas and a 3 m × 2 mm column packed with molecular sieve 13X 80-100. The gas flow was set to 20 mL min−1. The oven was operated isothermally at 100 °C. The gases were detected using a thermal conductivity detector (Varian) operated at 150 °C. Calibrations were performed by the injection of known quantities of pure oxygen diluted in a head space vial containing the same volume of solvent as used for measurements.Acknowledgements
Funding was provided by the Swiss National Science Foundation (SNSF Professorship PP00P2_133483/1) and by the University of Zurich. We thank Dr Gian Luca and Prof. Dr Jan-Dierk Grunwaldt for XAS measurement time as well as Dr Frank Krumeich (EMEZ, ETHZ) for TEM analyses. Beat Aeschlimann (ETHZ) is acknowledged for elemental analysis and we thank Dr Eric Reusser (ETHZ) for electron microprobe analyses. We thank Prof. Dr Ying Zhou (Southwest Petroleum University, Chengdu) and Prof. Dr Holger Dau (FU Berlin, Germany) for helpful discussions.References
- (a) R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS; (b) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; (c) R. Schlögl, ChemSusChem, 2010, 3, 209 CrossRef; (d) M. Kitano and M. Hara, J. Mater. Chem., 2010, 20, 627 RSC; (e) F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS; (f) H. G. Park and J. K. Holt, Energy Environ. Sci., 2010, 3, 1028 RSC; (g) W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966 CrossRef CAS; (h) D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231 RSC; (i) T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268 RSC; (j) S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. W. J. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74 CrossRef CAS.
- (a) H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS; (b) F. Zhang, C. W. Cady, G. W. Brudvig and H. J. M. Hou, Inorg. Chim. Acta, 2011, 366, 128 CrossRef CAS; (c) R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892 CrossRef CAS; (d) M. Risch, K. Klingan, J. Heidkamp, D. Ehrenberg, P. Chernev, I. Zaharieva and H. Dau, Chem. Commun., 2011, 47, 11912 RSC; (e) M. Wiechen, H.-M. Berends and P. Kurz, Dalton Trans., 2012, 41, 21 RSC; (f) P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871 CrossRef CAS.
- (a) F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018 RSC; (b) Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS; (c) D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467 CrossRef CAS; (d) V. B. R. Boppana and F. Jiao, Chem. Commun., 2011, 47, 8973 RSC; (e) T. Takashima, K. Hashimoto and R. Nakamuara, J. Am. Chem. Soc., 2012, 134, 1519 CrossRef CAS.
- (a) M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233 CrossRef CAS; (b) D. Shevela, S. Koroidov, M. M. Najafpour, J. Messinger and P. Kurz, Chem.–Eur. J., 2011, 17, 5415 CrossRef CAS.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nature Chem., 2011, 3, 461 CAS.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 2 CrossRef.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109 RSC.
- I. Zaharieva, M. Mahdi Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Energy Environ. Sci., 2011, 4, 2400 CAS.
- G. F. Swiegers, J. K. Clegg and R. Stranger, Chem. Sci., 2011, 2, 2254 RSC.
- (a) Q. Zhang, X. Liu, W. Fan and Y. Wang, Appl. Catal., B, 2011, 102, 207 CrossRef CAS; (b) S. Todorova, H. Kolev, J. P. Holgado, G. Kadinov, C. Bonev, R. Pereníguez and A. Caballero, Appl. Catal., B, 2010, 94, 46 CrossRef CAS; (c) T. E. Feltes, L. Espinosa-Alonso, E. de Smit, L. D'Souza, R. J. Meyer, B. M. Weckhuysen and J. R. Regalbuto, J. Catal., 2010, 270, 95 CrossRef CAS.
- (a) H. Bordeneuve, S. Guillemet-Fritsch, A. Rousset, S. Schuurman and V. Poulain, J. Solid State Chem., 2009, 182, 396 CrossRef CAS; (b) P. Mahata, D. Sarma, C. Madhu, A. Sundaresen and S. Natarajan, Dalton Trans., 2011, 40, 1952 RSC; (c) S. L. Cheng, Y. H. Tsai, K. M. Kuo, G. Chern and J. G. Lin, IEEE Trans. Magn., 2011, 47, 4325 CrossRef CAS.
- (a) F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS; (b) H. Zhang, Z. Zhan and X. Liu, J. Power Sources, 2011, 196, 8041 CrossRef CAS; (c) M. Mahmoud, T. A. Gad-Allah, K. M. El-Khatib and F. El-Gohary, Bioresour. Technol., 2011, 102, 10459 CrossRef CAS; (d) H. Liu, X. Zhu, M. Cheng, Y. Cong and W. Yang, Chem. Commun., 2011, 47, 2378 RSC.
- D. Reinen and O. Schmitz-DuMont, Z. Anorg. Allg. Chem., 1961, 312, 121 CrossRef CAS.
- (a) H. Xue, Z.-H. Li and L.-J. Zhu, Chinese J. Struct. Chem., 2010, 29, 1828 CAS; (b) K. Ikarashi, J. Sato, H. Kobayashi, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2002, 106, 9048 CrossRef CAS.
- (a) T. Hisatomi, K. Miyazaki, K. Takanabe, K. Maeda, J. Kubota, Y. Sakata and K. Domen, Chem. Phys. Lett., 2010, 486, 144 CrossRef CAS; (b) A. Bienholz, R. Blume, A. Knop-Gericke, F. Girgsdies, M. Behrens and P. Claus, J. Phys. Chem. C, 2011, 115, 999 CrossRef CAS; (c) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS; (d) C. C. Hu, Y. L. Lee and H. S. Teng, J. Phys. Chem. C, 2011, 115, 2805 CrossRef CAS.
- (a) I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358 RSC; (b) G. Buhler, A. Zharkouskaya and C. Feldmann, Solid State Sci., 2008, 10, 461 CrossRef; (c) F. Conrad, Y. Zhou, M. Yulikov, K. Hametner, S. Weyeneth, G. Jeschke, D. Günther, J.-D. Grunwaldt and G. R. Patzke, Eur. J. Inorg. Chem., 2010, 2036 CAS.
- (a) M. Rajamanthi and R. Seshadri, Curr. Opin. Solid State Mater. Sci., 2002, 6, 337 CrossRef; (b) F. Gao, Q. Lu, X. Meng and S. Komarneni, J. Mater. Sci., 2007, 43, 2377 CrossRef; (c) K. L. Harrison and A. Manthiram, Inorg. Chem., 2011, 50, 3613 CrossRef CAS.
- Y. Zhou, K. Vuille, A. Heel, B. Probst, R. Kontic and G. R. Patzke, Appl. Catal., A, 2010, 375, 140 CrossRef CAS.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841 CrossRef CAS.
- (a) V. di Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117 CrossRef CAS; (b) J. Töpfer, A. Feltz, D. Gräf, B. Hackl, L. Raupach and P. Weissbrodt, Phys. Status Solidi A, 1992, 134, 405 CrossRef.
- (a) A. Nakatsuka, Y. Ikeda, N. Nakayama and T. Mizota, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i109 Search PubMed; (b) P. Garcia Casado and I. Rasines, Z. Kristallogr., 1982, 160, 33 CrossRef CAS.
- T. Ressler, J. Phys. IV, 1997, C2–7, 269 Search PubMed.
- (a) B. Zapata, M. A. Valenzuela, P. Bosch, G. Fetter, I. Córdova, S. O. Flores and A. Vázquez, J. Metastable Nanocryst. Mater., 2004, 20–21, 163 CrossRef CAS; (b) S. K. Tripathy, M. Christy, N.-H. Park, E.-K. Suh, S. Anand and Y.-T. Yu, Mater. Lett., 2008, 62, 1006 CrossRef CAS.
- N. W. Ashcroft, N. D. Mermin, Festkörperphysik, Oldenbourg, 2001.
- S. Todorova, A. Naydenov, H. Kolev, K. Tenchev, G. Ivanov and G. Kadinov, J. Mater. Sci., 2011, 46, 7152 CrossRef CAS.
- (a) B. J. Tan, K. J. Klabunde and M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855 CrossRef CAS; (b) H. T. Zhang and X. H. Chen, Nanotechnology, 2006, 17, 1382 Search PubMed.
- ASTM Guide E. 1523-03, Standard Guide to Charge Control and Charge Referencing Techniques in X-ray Photoelectron Spectroscopy, ASTM International: West Conshohocken, PA, 2003.
Footnote |
| † Electronic Supplementary Information (ESI) available: PXRD and neutron diffraction data, TG/DSC curves, SEM images, XPS spectra, EXAFS investigations, catalytic data. See DOI: 10.1039/c2ra20169k/ |
| This journal is © The Royal Society of Chemistry 2012 |