A quick and efficient route to substituted quinolines by electrophilic cyclization of 1-(2-aminoaryl)-2-yn-1-ols†‡
Maddi Sridhar
Reddy
*,
Nuligonda
Thirupathi
and
Yalla Kiran
Kumar
Medicinal and Process Chemistry Division, Central Drug Research Institute, CSIR, Lucknow, 226 001, India. E-mail: sridharreddymaddi@yahoo.com; msreddy@cdri.res.in
First published on 14th March 2012
Abstract
A practical method for the synthesis of substituted quinolines from 2-aminoarylketones (via 1-(2-aminoaryl)-2-yn-1-ols) using mild and simple reaction conditions is described. A study of several electrophiles in various reaction conditions is presented. Out of three electrophilic iodine sources (I2, NIS and ICl) studied, I2 was found to work efficiently for the synthesis of 3-iodoquinolines. Several Brønsted acids (pTSA, PPTS, AcOH, TFA, HCl) were able to catalyze the formation of 2,4-substituted quinolines from the same starting materials. Good to excellent overall yields were observed with both aromatic and aliphatic substitutions at the reaction center.
Introduction
The wide occurrence of quinolines and their derivatives in nature, and their interesting biological and pharmacological activities have been continuously attracting synthetic, biological and medicinal chemists over the years. Though a number of methods are available for the synthesis of the quinoline motifs,1 due to their pharmaceutical importance,2 the development of new synthetic approaches for novel substitution patterns remains an active research area. In particular, the synthesis of halogen-substituted quinolines1b–e has attracted attention in recent chemistry because the halogen gives an excellent lever for further convenient structural elaborations.As part of a medicinal chemistry project in our laboratory, it was required to synthesize 3-iodoquinoline intermediates, and we were attracted by the recent publications of Larock,1b–c Flynn1d and Liang.1e Larock's method1b–c involves the electrophilic cyclization of aryl substituted propargylamines. This method works very well for 2,4-aryl substituted quinolines, however no reactions or unsatisfactory yields were observed for alkyl substitutions. On the other hand, Flynn et al.1d reported an elegant approach via electrophilic iodo cyclization of 1-(2-N,N-dimethylaminoaryl)-2-yn-ols (Scheme 1, eq 1). To improve its practicality and substrate scope, Liang et al.1e reported an improved method by substituting the dimethylamino group in Flynn's starting material with a tosylamino group (Scheme 1, eq 2). Although it works well for diaryl substituted substrates at pro-2C and pro-4C (numbering based on product), this method still suffers from multiple steps, long reaction times, elevated reaction temperatures, incompatible reaction conditions for acid sensitive substrates,3 and lower overall yields if one of the aryl groups (at pro-2C or pro-4C) is substituted by an alkyl group in the starting substrates. Furthermore, it did not work for dialkyl (at pro-2C and pro-4C) substrates even after prolonged reaction times in the given conditions (Scheme 1, eq 3).4 We were curious to know why unsubstituted amino (–NH2 instead of –NMe2 or –NHTs) starting materials, which were recently disclosed by Gabriele,1h were not used in Flynn's and Liang's approach to get the target products directly. This might avoid multiple steps and, would increase the reactivity of substrates and reduce reaction temperatures and times because of the steric bulkiness around the nitrogen is reduced (compared to Flynn's substrate) and nucleophilicity of the amino group is increased (compared to Liang's substrate). This might also allow access to the 1,3-dialkyl counterparts in the ultimate dehydration step, which in this case could be easier compared to the dehydroxylation followed by detosylation in Liang's approach.
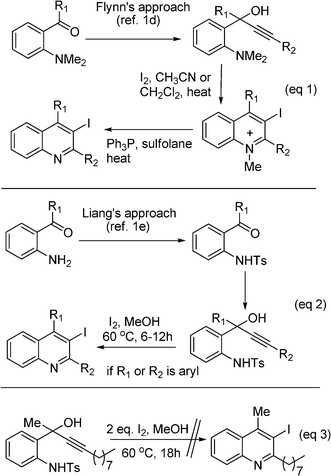 | ||
| Scheme 1 Analysis of previous approaches. | ||
Results and discussion
First we wanted to examine the reaction substrates with aliphatic groups at pro-2C and pro-4C which has been proved to be difficult in previous reports.1b–e Thus, we treated crude substrate 2a,1g,5 prepared from 1a and 1-hexynylmagnesium bromide, with 2 equivalents of I2 in MeOH. Within ten minutes at room temperature the starting material disappeared (TLC) and two products were found. The major product was identified as the expected product 3a and the minor one as 3,6-diiodoquinoline 4 (see Table 1). The latter was formed through electrophilic substitution followed by electrophilic cyclization as the reverse is very difficult, and hence the ratio of the products remained constant even after extended reaction times. Reaction of 2a with other electrophilic iodine sources such as N-iodosuccinimide (NIS) and ICl also led to the formation of a mixture of 3a and 4 but in a different ratio. The use of NaHCO3 along with I2 in MeOH gave a slightly better result but 4 was still formed in 10% yield.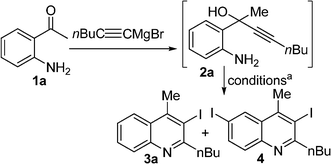
| Entry | I+ source | Base | Solvent | Temp. | Time (min) | Yieldb | |
|---|---|---|---|---|---|---|---|
| 3a | 4 | ||||||
| a The reactions were carried out with 5 mmol substrate using 2 eq. I2 at 0.13 M concentration. b Isolated overall yields after two steps. | |||||||
| 1 | I2 | — | MeOH | rt | 10 | 62 | 19 |
| 2 | NIS | — | MeOH | rt | 10 | 7 | 71 |
| 3 | ICl | — | MeOH | rt | 10 | 13 | 52 |
| 4 | I2 | NaHCO3 | MeOH | rt | 10 | 66 | 10 |
| 5 | NIS | NaHCO3 | MeOH | rt | 10 | 22 | 55 |
| 6 | ICl | NaHCO3 | MeOH | rt | 10 | 18 | 50 |
| 7 | I2 | — | CH2Cl2 | rt | 20 | 74 | trace |
| 8 | NIS | — | CH2Cl2 | rt | 20 | 48 | 28 |
| 9 | ICl | — | CH2Cl2 | rt | 20 | 52 | 18 |
NIS and ICl, using the same base and solvent, also resulted in a mixture of products. However, a change of solvent from MeOH to CH2Cl2 cleanly produced 3a as the sole product by using 2 eq. I2 and no base. Even in CH2Cl2, NIS and ICl produced a mixture of products though in favour of the required iodoquinoline. Meanwhile, we also performed the optimization with substrate 2b in parallel and the results are described in Table 2. Surprisingly, 2b, on exposure to I2 in MeOH at ambient temperature, gave quinoline 5b as a byproduct.6 By switching the solvent from MeOH to CH2Cl2, the byproduct 5b was formed in negligible quantities (8%). In this case, we did not observe any aromatic electrophilic substitution product. We reasoned that the HI formed during the electrophilic iodo cyclization could be responsible for the formation of the non-iodinated product 5.6 We conclude that the activation energy of electrophilic cyclizations is considerably reduced with the aromatic substitution at pro-2C, resulting in no formation of the diiodoquinoline derivative like in the case of 1a. On the other hand, in the case of pro-2C alkyl substrate 2a, HI-induced electrophilic cyclization was not favored due to absence of conjugation and hence the non-iodinated quinoline derivative byproduct was not observed (Table 1).
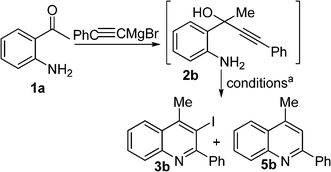
| Entry | I+ source | Base | Solvent | Temp. | Time (min) | Yieldb | |
|---|---|---|---|---|---|---|---|
| 3b | 5b | ||||||
| a The reactions were carried out with 5 mmol substrate using 2 eq. I2 at 0.13 M concentration. b Isolated overall yields after two steps. | |||||||
| 1 | I2 | — | MeOH | rt | 5 | 35 | 30 |
| 2 | I2 | NaHCO3 | MeOH | rt | 5 | 46 | 20 |
| 3 | I2 | — | CH2Cl2 | rt | 10 | 67 | 8 |
| 4 | I2 | NaHCO3 | CH2Cl2 | rt | 10 | 68 | 9 |
With the optimal conditions (2 eq. of I2 in CH2Cl2 at room temperature for 10–20 min) in hand, the scope of the approach was examined. Several 1-(2-aminoaryl)-2-yn-1-ols were prepared and tested for their reactivity, and the results are presented in Table 3. Dialkyl substrates 2c–2g reacted well under the standard conditions to give 70–81% overall yields. Substrates 2h–2k bearing aromatic substituents were more reactive and were smoothly converted into the corresponding products in 67–92% overall yields.
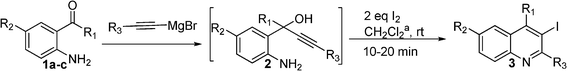
| Entry | 1 | 2 | Time (min) | Product | Yieldb |
|---|---|---|---|---|---|
| a The reactions were carried out at 0.13 M concentration. b Isolated overall yields after two steps. | |||||
| 1 | 1a | 2a | 20 | 3a | 74% |
| 2 | 1a | 2b | 10 | 3b | 67% |
| 3 | 1a |
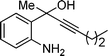 2c
2c
|
20 |
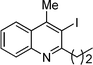 3c
3c
|
75% |
| 4 | 1a |
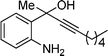 2d
2d
|
20 |
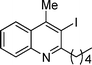 3d
3d
|
78% |
| 5 | 1a |
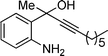 2e
2e
|
20 |
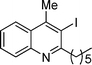 3e
3e
|
81% |
| 6 | 1a |
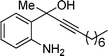 2f
2f
|
20 |
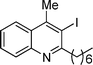 3f
3f
|
80% |
| 7 | 1a |
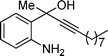 2g
2g
|
10 |
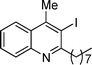 3g
3g
|
70% |
| 8 |
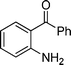 1b
1b
|
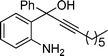 2h
2h
|
10 |
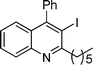 3h
3h
|
74% |
| 9 | 1b |
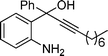 2i
2i
|
10 |
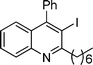 3i
3i
|
81% |
| 10 | 1b |
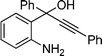 2j
2j
|
10 |
 3j
3j
|
69% |
| 11 |
 1c
1c
|
 2k
2k
|
10 |
 3k
3k
|
92% |
| 12 | 1a |
 2l
2l
|
20 |
 3l
3l
|
63% |
Notably, pro-4C-aryl substrates gave relatively higher yields compared to pro-2C-aryl substrates. It is worth mentioning that the presence of a withdrawing group on the aromatic ring, as in case of 2k, did not have a negative impact on reactivity, with product 3k being formed in 92% yield. Similarly, substrate 2l, bearing the acid sensitive OTHP group, also provided the corresponding quinoline 5l, though in a moderate overall yield of 63%. We also performed the reaction of 2d to 3d on a 50 mmol scale (6.7 g of 1a) which produced the product in almost the same percentage yield as the experiment that was carried out at a smaller scale (11.7 g, 70% instead of 78%).
A plausible mechanism for electrophilic iodocyclization is presented in Scheme 2. Electrophilic iodine initiated cyclization could lead to the intermediate A which on dehydration/aromatization forms the 3-iodoquinoline 3.
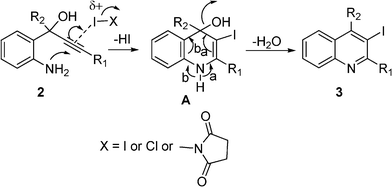 | ||
| Scheme 2 A plausible mechanism for the formation of iodoquinolines. | ||
As we have seen, the results obtained with substrate 2b (Table 2) suggested that Brønsted acid might catalyze the formation of 2,3-substituted quinolines (pro-3H counterparts). We accordingly tested the reactivity of 2b under acidic conditions. We began our study by treating 2b with HCl, AcOH, TFA, TfOH, pTSA, and PPTS under various conditions; the results are summarized in Table 4. As can be seen from Table 4, almost all catalysts worked very well, with the exception of TfOH. AcOH and TFA were found to work at room temperature to give 62% and 55% overall yields respectively. PPTS produced a better yield (78%) though it needed a higher temperature (60 °C).

| Entry | 2 | Reagent (5 mmol%) | Temp. (°C) | Time (h) | 5 | Overall yieldb |
|---|---|---|---|---|---|---|
| a All reactions were carried out with 5 mmol substrate using 5 mol% catalyst in MeOH (0.13 M). b Isolated overall yields after two steps. | ||||||
| 1 | 2b | HCl | 50 | 2 | 5b | 60% |
| 2 | 2b | AcOH | rt | 5 | 5b | 62% |
| 3 | 2b | TFA | rt | 1 | 5b | 55% |
| 4 | 2b | TfOH | rt | 1 | 5b | 30% |
| 5 | 2b | pTSA | 50 | 5 | 5b | 70% |
| 6 | 2b | PPTS | 50 | 1 | 5b | 78% |
To explore the feasibility of the method, various substrates bearing alkyl and/or aryl substituents were subjected to the reaction and the results are reported in Table 5. As is apparent from Table 4, the method worked very well for both aliphatic and aromatic substituted substrates giving excellent overall yields (70–90%). The same conversion was reported earlier by Gabriele et al. using Cu and Pd metal catalysts.1h Although the method proposed here appears to be less general and less versatile with respect with the Gabriele's method, it nevertheless shows that acid catalysis is also possible for the conversion of substrates 2a–j in to quinolines 5a–j.
| Entry | 2 | Time (h) | 5 | Overall yieldb |
|---|---|---|---|---|
| a All reactions were carried out with 5 mmol substrate using 5 mol% catalyst in MeOH (0.13 M) at 50 °C. b Isolated overall yields after two steps. | ||||
| 1 | 2a | 5 |
 5a
5a
|
85% |
| 2 | 2b | 1 |
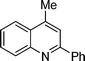 5b
5b
|
78% |
| 3 | 2c | 5 |
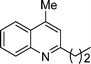 5c
5c
|
74% |
| 4 | 2d | 5 |
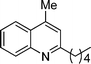 5d
5d
|
76% |
| 5 | 2e | 5 |
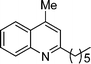 5e
5e
|
90% |
| 6 | 2f | 5 |
 5f
5f
|
70% |
| 7 | 2g | 5 |
 5g
5g
|
70% |
| 8 | 2h | 2 |
 5h
5h
|
78% |
| 9 | 2i | 2 |
 5i
5i
|
80% |
| 10 | 2j | 1 |
 5j
5j
|
80% |
Conclusion
In summary, an efficient and convergent approach has been described for the synthesis of conveniently substituted quinolines through an electrophilic cyclization of 1-(2-aminoaryl)-2-yn-1-ols. The method features mild conditions and good overall yields in addition to benign substitution compatibility.Experimental section
General information
All reagents were commercial and were used without further purification unless otherwise noted. Infrared spectra were recorded with FT-IR as a thin film and are expressed in cm−1. 1H NMR (at 200 or 300 MHz) and 13C NMR (50 or 75 MHz) spectra were recorded using CDCl3 as solvents and TMS as an internal standard. Mass spectra were obtained on a ESI mass spectrometer and HR/ESI mass spectra were obtained on a high resolution ESI mass spectrometer.2-Butyl-3-iodo-4-methylquinoline (3a). 74% over all yield (in two steps); light yellow solid; m.p. 75–78 °C; Rf = 0.75 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.04-7.98 (m, 2H), 7.72-7.64 (m, 1H), 7.53–7.45 (m, 1H), 3.27–3.20 (m, 2H), 2.91 (s, 3H), 1.86–1.74 (m, 2H), 1.58–1.46 (m, 2H), 1.00 (t, 3H, J = 7.3 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.8, 148.3, 146.7, 129.6, 129.4, 126.8, 126.5, 124.3, 102.3, 43.3, 31.4, 25.7, 22.9, 14.1. ; IR (KBr) ν: 3020, 2959, 2866, 1636, 1571, 1492, 1459, 1216, 955, 668, 559 cm−1; HRMS (ESI) m/z calcd for C14H17IN [M + H]+: 326.0406, found: 326.0739.
3-Iodo-4-methyl-2-phenylquinoline (3b). 67% over all yield (in two steps); known compound;1e light yellow solid, m.p. 105–108 °C; (lit.1e 105–110 °C); Rf= 0.65 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.15–8.08 (m, 2H), 7.78–7.70 (m, 2H), 7.62–7.45 (m, 6H), 3.00 (s, 3H); 13C NMR (50 MHz, CDCl3) δ: 162.1, 149.2, 146.6, 144.4, 130.2, 129.7, 129.2, 128.6, 128.1, 127.3, 124.4, 100.6, 26.0 ; IR (KBr) ν: 2969, 2852, 1634, 1443, 1344, 1219, 771, 691, 547, 493 cm−1; HRMS (ESI) m/z calcd for C16H13IN [M + H]+: 346.0093, found: 346.0162.
3-Iodo-4-methyl-2-propylquinoline (3c). 75% over all yield (in two steps); light yellow solid; m.p. 85–88 °C; Rf = 0.73 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.04–7.98 (m, 2H), 7.72–7.65 (m, 1H), 7.54–7.46 (m, 1H), 3.25–3.17 (m, 2H), 2.91 (s, 3H), 1.92–1.79 (m, 2H), 1.08 (t, 3H, J = 7.3 Hz); 13C NMR (75 MHz, CDCl3) δ: 162. 6, 148.2, 146.7, 129.6, 129.5, 126.9, 126.5, 124.3, 102.4, 45.5, 25.68, 22.6, 14.2; IR (KBr) ν: 2957, 2928, 2867, 1638, 1491, 1459, 1379, 966, 772, 747, 658, 558 cm−1; HRMS (ESI) m/z calcd for C13H15IN [M + H]+: 312.0249, found: 312.0378.
3-Iodo-4-methyl-2-pentylquinoline (3d). 78% over all yield (in two steps); light yellow solid; m.p. 72–75 °C; Rf = 0.70 (EtOAc/hexanes = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.06–7.97 (m, 2H), 7.72–7.64 (m, 1H), 7.53–7.45 (m, 1H), 3.27–3.18 (m, 2H), 2.90 (s, 3H), 1.88–1.76 (m, 2H), 1.54–1.34 (m, 4H), 0.94 (t, 3H, J = 7.0 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.8, 148.3, 146.7, 129.6, 129.5, 126.9, 126.5, 124.3, 102.4, 43.6, 32.0, 29.0, 25.7, 22.7, 14.3; IR (KBr) ν: 3067, 2928, 2860, 1640, 1568, 1458, 1379, 962, 758, 657 cm−1; HRMS (ESI) m/z calcd for C15H18IN [M + H]+: 340.0562, found: 340.0563.
2-Hexyl-3-iodo-4-methylquinoline (3e). 81% over all yield (in two steps); orange oil; Rf = 0.67 (EtOAc/hexanes = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.04–7.98 (m, 2H), 7.73–7.65 (m, 1H), 7.53–7.46 (m, 1H), 3.27–3.18 (m, 2H), 2.91 (s, 3H), 1.88–1.74 (m, 2H), 1.56–1.30 (m, 6H), 0.91 (t, 3H, J = 6.8 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.8, 148.2, 146.7, 129.5, 129.5, 126.8, 126.4, 124.3, 102, 7, 43.6, 31.8, 29.5, 29.2, 25.7, 22. 8, 14.2; IR (KBr) ν: 2928, 2860, 2364, 1629, 1488, 1449, 1378, 1218, 966, 764, 672, 532 cm−1; HRMS (ESI) m/z calcd for C16H21IN [M + H]+: 354.0719, found: 354.0782.
2-Heptyl-3-iodo-4-methylquinoline (3f). 80% over all yield (in two steps); orange oil; Rf = 0.62 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.04–7.97 (m, 2H), 7.72–7.64 (m, 1H), 7.53–7.46 (m, 1H), 3.26–3.18 (m, 2H), 2.90 (s, 3H), 1.88–1.74 (m, 2H), 1.54–1.24 (m, 8H), 0.89 (t, 3H, J = 6.6 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.8, 148.2, 146.7, 129.5, 129.4, 126.8, 126.4, 124.3, 102.3, 43.60, 32.0, 29.8, 29.3, 25.7, 22.8, 14.3; IR (KBr) ν: 2926, 2856, 2363, 1637, 1460, 1379, 769, 668, 515 cm−1; HRMS (ESI) m/z calcd for C17H23IN [M + H]+: 368.0875, found: 368.1523.
3-Iodo-4-methyl-2-octylquinoline (3g). 70% over all yield (in two steps); yellow oil; Rf = 0.60 (EtOAc/hexanes = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.05–7.98 (m, 2H), 7.73–7.65(m, 1H), 7.54–7.46 (m, 1H), 3.27–3.18 (m, 2H), 2.91 (s, 3H), 1.87–1.75 (m, 2H), 1.54–1.20 (m, 10H), 0.88 (t, 3H, J = 6.9 Hz); 13C NMR (75 MHz, CDCl3) δ: 162.8, 148.3, 146.7, 129.6, 129.5, 126.9, 126.5, 124.4, 102.4, 43.6, 32.0, 29.8, 29.6, 29.4, 29.3, 25.7, 22.8, 14.3; IR (KBr) ν: 2924, 2856, 1627, 1454, 1377, 1257, 960, 766, 658, 551 cm−1; HRMS (ESI) m/z calcd for C18H25IN [M + H]+: 382.1032, found: 382.1395.
3-Iodo-2-pentyl-4-phenylquinoline (3h). 74% over all yield (in two steps); yellow solid; m.p. 85–90 °C; Rf = 0.9 (EtOAc/hexane 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.07 (d, 1H, J = 8.25 Hz), 7.72–7.64 (m, 1H), 7.60–7.50 (m, 3H), 7.37–7.19 (m, 4H), 3.28 (t, 2H, J = 7.7 Hz), 1.95–1.80 (m, 2H), 1.58–1.38 (m, 4H), 0.95 (t, 3H, J = 6.9 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.9, 154.0, 147.0, 142.3, 129.8, 129.2, 128.8, 128.7, 128.4, 127.1, 126.9, 126.5, 100.4, 43.3, 32.0, 29.0, 22.7, 14.2; IR (KBr) ν: 3063, 2951, 2860, 1623, 1572, 1449, 1379, 1218, 699, 610, 519 cm−1; HRMS (ESI) m/z calcd for C20H21IN [M + H]+: 402.0719, found: 402.0730.
2-Hexyl-3-iodo-4-phenylquinoline (3i). 81% over all yield (in two steps); yellow solid; m.p. 80–85 °C; Rf = 0.85 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.06 (d, 1H, J = 8.4 Hz), 7.72–7.64 (m, 1H), 7.60–7.50 (m, 3H), 7.37–7.19 (m, 4H), 3.34–3.22 (m, 2H), 1.94–1.80 9m, 2H), 1.60–1.28 (m, 6H), 0.91 (t, 3H, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3) δ: 162.9, 154.0, 147.0, 142.4, 129.8, 129.20, 128.8, 128.7, 128.4, 127.1, 126.9, 126.5, 100.4, 43.4, 31.8, 29.5, 29.3, 22.8, 14.2; IR (KBr) ν: 2926, 2722, 1633, 1467, 1218, 1111, 1030, 769, 700, 612 cm−1; HRMS (ESI) m/z calcd for C21H23IN [M + H]+: 416.0875, found: 416.0884.
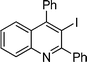
3-Iodo-2,4-diphenylquinoline (3j). 69% over all yield (in two steps); known compound;1e yellow solid; m.p. 115–118 °C; (lit.1e 118–122 °C); Rf= 0.55 (EtOAc/hexanes = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.19–8.13 (d, 1H, J = 8.3 Hz), 7.77–7.38 (m, 11H), 7.34–7.24 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 162.0, 154.8, 147.0, 143.9, 142.3, 130.2, 129.5, 129.4, 129.2, 128.7, 128.7, 128.6, 128.1, 127.5, 127.4, 127.0, 98.6; IR (KBr) ν: 1636, 1480, 1442, 1340, 1220, 771, 697, 625, 583 cm−1; HRMS (ESI) m/z calcd for C21H15IN [M + H]+ 408.0249, found: 408.0519.
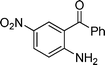
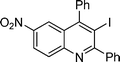
3-Iodo-6-nitro-2-pentyl-4-phenylquinoline (3k). 92% over all yield (in two steps); yellow solid; m.p. 100–105 °C; Rf = 0.7 (EtOAc/hexane 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.44 (dd, 1H, J = 9.2, 2.4 Hz), 8.27 (d, 1H, J = 2.4 Hz), 8.17 (d, 1H, J = 9.2 Hz), 7.65–7.55 (m, 3H), 7.28–7.20 (m, 2H), 3.31 (t, 2H, J = 7.8 Hz), 1.98–1.84 (m, 2H), 1.60–1.20 (m, 6H), 0.97 (t, 3H, J = 7.0 Hz); 13C NMR (75 MHz, CDCl3) δ 167.1, 155.8, 149.0, 145.5, 140.8, 130.7, 129.3, 129.1, 129.0, 125.8, 123.7, 123.2, 102.9, 43.7, 31.9, 28.6, 22.6, 14.2; IR (KBr) ν: 3015, 2926, 2857, 1622, 1528, 1343, 1219, 840, 768, 697, 611 cm−1; HRMS (ESI) m/z calcd for C20H20IN2O2 [M + H]+ 447.0569, found: 447.0605.
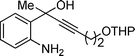
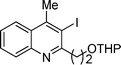
3-Iodo-4-methyl-2-(2-(tetrahydro-2H-pyran-2-yloxy)ethyl)quinoline (3l). 63% over all yield (in two steps); yellow oil; Rf = 0.4 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.04–7.97 (m, 2H), 7.72–7.64 (m, 1H), 7.52–7.46 (m, 1H), 4.77–4.73 (m, 1H), 4.32-4.31 (m, 1H), 4.03–3.84 (m, 2H), 3.62–3.47 (m, 3H), 2.90 (s, 3H), 1.90–1.44 (m, 6H); 13C NMR (75 MHz, CDCl3) δ: 159.5, 148.2, 146.6, 129.6, 129.6, 127.0, 126.7, 124.3, 102.7, 99.0, 66.5, 62.3, 43.2, 30.8, 29.8, 25.6, 19.6 ; IR (KBr) ν: 2940.6, 1636, 1448, 1378, 1125, 1026, 758, 635, 578 cm−1; ESI-MS m/z :M+ 397.9.
2-Butyl-3,6-diiodo-4-methylquinoline (4). yellow solid; m.p. 78–81 °C; Rf = 0.8 (EtOAc/hexane = 1/9); 1H NMR (300 MHz, CDCl3) δ: 8.34 (s, 1H), 7.88 (d, 1H, J = 8.8 Hz), 7.70 (d, 1H, J = 8.8 Hz), 3.19 (t, 2H, J = 7.7 Hz), 2.82 (s, 3H), 1.82–1.72 (m, 2H), 1.57–1.42 (m, 2H), 0.99 (t, 3H, J = 7.3 Hz); 13C NMR (75 MHz, CDCl3) δ: 163.5, 147.1, 145.7, 138.3, 133.4, 131.2, 128.4, 103.2, 92.1, 43.3, 31.18, 25.7, 22.9, 14.1; ESI-MS m/z: M+ 452.1.
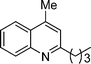
2-Butyl-4-methylquinoline (5a). 78% over all yield (in two steps); known compound;1h yellow oil; Rf = 0.8 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.04 (d, 1H, J = 8.2 Hz), 7.95 (d, 1H, J = 7.8 Hz), 7.70–7.63 (m, 1H), 7.54–7.46 (m, 1H), 7.14 (s, 1H), 2.93 (t, 2H, J = 7.7 Hz), 2.68 (s, 3H), 1.86–1.73 (m, 2H), 1.52–1.38 (m, 2H), 0.96 (t, 3H, J = 7.3 Hz); 13C NMR (75 MHz, CDCl3) δ: 162.8, 147.8, 144.1, 129.4, 129.0, 126.8, 125.4, 123.6, 122.1, 39.0, 32.2, 22.8, 18.7, 14.0; IR (KBr) ν: 2956, 2929, 2865, 1606, 1563, 1507, 1448, 1350, 863, 758, 663, 537, 500 cm−1; HRMS (ESI) m/z calcd for C14H18N [M + H]+: 200.1439, found: 200.1474.
4-Methyl-2-phenylquinoline (5b). 85% over all yield (in two steps); known compound;11 yellow oil ; Rf = 0.72 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.22–8.12 (m, 3H), 8.01 (d, 1H, J = 7.8 Hz), 7.77–7.68 (m, 2H), 7.60–7.42 (m, 4H), 2.78 (s, 3H); 13C NMR (50 MHz, CDCl3) δ: 157.1, 148.2, 144.8, 139.8, 130.3, 129.4, 129.2, 128.8, 127.6, 127.3, 126.0, 123.6, 119.8, 19.0; IR (KBr) ν: 3066, 2926, 1602, 1552, 1449, 1348, 1219, 865, 766, 694, 573, 538 cm−1; HRMS (ESI) m/z calcd for C16H14N [M + H]+: 220.1126, found: 220.1161.
4-Methyl-2-propylquinoline (5c). 74% over all yield (in two steps); known compound;7 clear liquid; Rf = 0.83 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.05 (d, 1H, J = 8.3 Hz), 7.94 (d, 1H, J = 7.8 Hz), 7.70–7.63 (m, 1H), 7.56–7.45 (m, 1H), 7.14 (s, 1H), 2.91 (t, 2H, J = 7.6 Hz), 2.68 (s, 3H), 1.90–1.77 (m, 2H), 1.02 (t, 3H, J = 7.3 Hz); 13C NMR (75 MHz, CDCl3) δ: 162.5, 147.7, 144.1, 129.3, 129.0, 126.8, 125.4, 123.5, 122.1, 41.2, 23.3, 18.6, 14.1; IR (KBr) ν: 3066, 2959, 2931, 2870, 1605, 1562, 1509, 1448, 1411, 1218, 861, 758, 664, 524 cm−1; HRMS (ESI) m/z calcd for C13H16N [M + H]+: 186.1283, found: 186.1314.
4-Methyl-2-pentylquinoline (5d). 74% over all yield (in two steps); known compound;7 clear liquid; Rf = 0.8 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.05 (d, 1H, J = 8.3 Hz), 7.94 (d, 1H, J = 8.1 Hz), 7.70–7.63 (m, 1H), 7.52–7.45 (m, 1H), 7.14 (s, 1H), 2.91 (t, 2H, J = 7.7 Hz), 2.67 (s, 3H), 1.87–1.70 (m, 2H), 1.48–1.32 (m, 4H), 0.90 (t, 3H, J = 6.73 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.8, 147.8, 144.2, 129.4, 129.0, 126.8, 125.4, 123.6, 122.1, 39.3, 31.9, 29.5, 22.7, 18.7, 14.1; IR (KBr) ν: 3014, 2930, 2861, 1606, 1509, 1452, 1412, 1216, 763, 667, 610, 479 cm−1; HRMS (ESI) m/z calcd for C15H20N [M + H]+: 214.1596, found: 214.1808.
2-Hexyl-4-methylquinoline (5e). 90% over all yield (in two steps); known compound;8 yellow oil; Rf = 0.76 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.02 (d, 1H, J = 8.1 Hz), 7.95 (d, 1H, J = 8.2 Hz), 7.70–7.63 (m, 1H), 7.53–7.46 (m, 1H, 7.14 (s, 1H), 2.92 (t, 2H, J = 7.8 Hz), 2.68 (s, 3H), 1.86–1.72 (m, 2H), 1.48–1.22 (m, 6H), 0.88 (t, 3H, J = 6.6 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.8, 147.8, 144.1, 129.4, 129.0, 126.8, 125.4, 123.6, 122.1, 39.3, 31.8, 30.1, 29.3, 22.6, 18.7, 14.1; IR (KBr) ν: 2927, 2857, 1605, 1562, 1509, 1449, 1411, 1349, 1218, 863, 761, 666, 594, 512 cm−1; HRMS (ESI) m/z calcd for C16H22N [M + H]+: 228.1752, found: 228.1882.
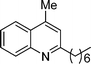
2-Heptyl-4-methylquinoline (5f). 70% over all yield (in two steps); known compound;9 yellow oil; Rf = 0.73 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.04 (d, 1H, J = 8.1 Hz), 7.95 (d, 1H, J = 7.8 Hz), 7.70–7.63 (m, 1H), 7.53–7.46 (m, 1H), 7.14 (s, 1H), 2.92 (t, 2H, J = 7.8 Hz), 2.68 (s, 3H), 1.90–1.70 (m, 2H), 1.50–1.20 (m, 8H), 0.87 (t, 3H, J = 6.5 Hz); 13C NMR (75 MHz, CDCl3) δ: 162.9, 147.8, 144.2, 129.4, 129.1, 126.9, 125.5, 123.7, 122.2, 39.4, 31.9, 30.2, 29.7, 29.3, 22.8, 18.8, 14.2; IR (KBr) ν: 2929, 2858, 1608, 1562, 1509, 1454, 1354, 1218, 868, 761, 667, 531 cm−1; HRMS (ESI) m/z calcd for C17H24N [M + H]+: 242.1909, found: 242.2139.
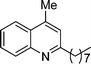
4-Methyl-2-octylquinoline (5g). 70% over all yield (in two steps); yellow oil; Rf = 0.7 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.04 (d, 1H, J = 8.3 Hz), 7.95 (d, 1H, J = 8.1 Hz), 7.70–7.63 (m, 1H), 7.54–7.46 (m, 1H), 7.14 (s, 1H), 2.92 (t, 2H, J = 7.7 Hz), 2.68 (s, 3H), 1.86–1.64 (m, 2H), 1.50–1.20 (m, 10H), 0.87 (t, 3H, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3) δ: 162.9, 147.9, 144.3, 129.5, 129.1, 126.9, 125.5, 123.7, 122.2, 39.4, 32.0, 30.2, 29.8, 29.6, 29.3, 22.8, 18.8, 14.2; IR (KBr) ν: 2926, 2856, 1635, 1561, 1508, 1452, 1373, 1218, 860, 767, 689, 658, 507 cm−1; HRMS (ESI) m/z calcd for C18H26N [M + H]+: 256.2065, found: 256.2148.
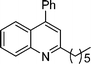
2-Pentyl-4-phenylquinoline (5h). 78% over all yield (in two steps); known compound;10 liquid; Rf = 0.7 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.12 (d, 1H, J = 8.17 Hz), 7.86 (d, 1H, J = 7.7 Hz), 7.72–7.65 (m, 1H), 7.57–7.40 (m, 6H), 7.25 (s, 1H), 3.00 (t, 2H, J = 7.7 Hz), 1.90–1.78 (m, 2H), 1.50–1.32 (m, 4H), 0.91 (t, 3H, J = 6.7 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.7, 148.6, 148.5, 138.3, 129.6, 129.2, 128.5, 128.3, 125.7, 125.7, 125.3, 121.6, 39.4, 31.9, 29.8, 22.6, 14.1; IR (KBr) ν: 3060, 2933, 2860, 1595, 1489, 1449, 1409, 1218, 884, 769, 701, 664, 577, 539 cm−1; HRMS (ESI) m/z calcd for C20H22N [M + H]+: 276.1752, found: 276.1864.
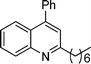
2-Hexyl-4-phenylquinoline (5i). 80% over all yield (in two steps); yellow oil; Rf = 0.7 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.12 (d, 1H, J = 8.3 Hz), 7.87 (d, 1H, J = 7.9 Hz), 7.73–7.64 (m, 1H), 7.57–7.40 (m, 6H), 7.25 (s, 1H), 3.00 (t, 2H, J = 7.7 Hz), 1.90–1.78 (m, 2H), 1.50–1.24 (m, 6H), 0.88 (t, 3H, J = 6.7 Hz); 13C NMR (50 MHz, CDCl3) δ: 162.7, 148.5, 138.4, 129.6, 129.3, 128.5, 128.3, 125.7, 125.6, 125.3, 121.6, 39.4, 31.8, 30.1, 29.8, 29.4, 22.6, 14.1; IR (KBr) ν: 3063, 2928, 2857, 1594, 1557, 1490, 1409, 1218, 881, 767, 701, 659, 615, 576 cm−1; HRMS (ESI) m/z calcd for C21H24N [M + H]+: 290.1909, found: 290.2009.
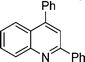
2,4-Diphenylquinoline (5j). 80% over all yield (in two steps); known compound;1h yellow solid m.p. 105–108 °C; (lit.1 107–108 °C); white solid; Rf = 0.62 (EtOAc/hexane = 2/8); 1H NMR (300 MHz, CDCl3) δ: 8.30–8.17 (m, 3H), 7.92 (d, 1H, J = 7.8 Hz), 7.83 (s, 1H), 7.78–7.70 (m, 1H), 7.63–7.44 (m, 9H); 13C NMR (50 MHz, CDCl3) δ: 156.9, 149.2, 148.9, 139.7, 138.4, 130.2, 129.6, 129.4, 128.9, 128.6, 128.5, 127.6, 126.4, 125.8, 125.7, 119.4; IR (KBr) ν: 3061, 2966, 1590, 1546, 1489, 1407, 1357, 1075, 1027, 883, 769, 697, 616, 590, 544 cm−1; HRMS (ESI) m/z calcd for C21H16N [M + H]+ 282.1283, found: 282.1464.
Acknowledgements
We thank Prof. Pierre Deslongchamps, University of Sherbrooke, Canada and Tushar K. Chakraborty, Director, CSIR-CDRI, India for their encouragement. NT and YKK thank CSIR for the award of fellowship. We thank SAIF division, CDRI for analytical support (NMR, IR, Mass).References
- Some recent examples: (a) A. K. Verma, T. Kesharwani, J. Singh, V. Tandon and R. C. Larock, Angew. Chem., Int. Ed., 2009, 48, 1138 CrossRef; (b) X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763–766 CrossRef CAS; (c) X. Zhang, T. Yao, M. A. Campo and R. C. Larock, Tetrahedron, 2005, 66, 1177–1187 CrossRef; (d) K. O. Hessian and B. L. Flynn, Org. Lett., 2006, 8, 243–246 CrossRef CAS; (e) S. Ali, H.-T. Zhu, X.-F. Xia, K.-G. Ji, Y.-F. Yang, X.-R. Song and Y.-M. Liang, Org. Lett., 2011, 13, 2598–2601 CrossRef CAS; (f) M. J. Sandelier and P. DeShong, Org. Lett., 2007, 9, 3209–3212 CrossRef CAS; (g) Z. Huo, I. D. Gridnev and Y. Yamamoto, J. Org. Chem., 2010, 75, 1266–1270 CrossRef CAS; (h) B. Gabriele, R. Mancuso, G. Salerno, G. Ruffolo and P. Plastina, J. Org. Chem., 2007, 72, 6873–6877 CrossRef CAS; (i) B. Jiang and Y.-G. Si, J. Org. Chem., 2002, 67, 9449–9451 CrossRef CAS; (j) B. Gabriele, R. Mancuso, G. Salerno, E. Lupinacci, G. Ruffolo and M. Costa, J. Org. Chem., 2008, 73, 4971–4977 CrossRef CAS . And the references therein.
- Some recent examples for pharmaceutical importance: (a) O. Billker, V. Lindo, M. Panico, A. E. Etienne, T. Paxton, A. Dell, M. Rogers, R. E. Sinden and H. R. Morris, Nature, 1998, 392, 289–292 CrossRef CAS; (b) S. W. Elmore, M. J. Coghlan, D. D. Anderson, J. K. Pratt, B. E. Green, A. X. Wang, M. A. Stashko, C. W. Lin, C. M. Tyree, J. N. Miner, P. B. Jacobson, D. M. Wilcox and B. C. Lane, J. Med. Chem., 2001, 44, 4481–4491 CrossRef CAS; (c) W. Peczynskaczoch, F. Pognan, L. Kaczmarek and J. Boratynski, J. Med. Chem., 1994, 37, 3503–3510 CrossRef CAS; (d) A. A. Joshi and C. L. Vishwanathan, Bioorg. Med. Chem. Lett., 2006, 16, 2613–2617 CrossRef CAS; (e) C. Portela, C. M. M. Afonso, M. M. Pinta and M. J. Ramos, Bioorg. Med. Chem., 2004, 12, 3313–3321 CrossRef CAS . And the references therein.
- We observed that acid sensitive groups like OTHP and OTBS could not withstand Liang's electrophilic cyclization conditions.
- The starting material has disappeared and a mixture of unknown products' spots were observed by TLC.
- 1H NMR of crude 2a showed a single product. In subsequent investigations, we found that purification of 2 did not show any considerable change in product yields. Also, 2 was observed to be sensitive to silica gel.
- In later observations, we found that 2b was slowly converted to quinoline 5b upon prolonged standing at room temperature.
- B. K. Mehta, K. Yanagisawa, M. Shiro and H. Kotsuki, Org. Lett., 2003, 5, 1605–1608 CrossRef CAS.
- F. Minisci, F. Fontana, G. Pianese and Y. M. Yan, J. Org. Chem., 1993, 58, 4207–4211 CrossRef CAS.
- G. I. Nikishin, L. L. Sokova, V. M. Makhaev and N. I. Kapustina, Russ. Chem. Bull., Int. Ed., 2007, 54, 1599–1605 Search PubMed.
- R. Martínez, D. J. Ramón and M. Yus, J. Org. Chem., 2008, 73, 9778–9780 CrossRef.
- R. Martínez, D. J. Ramón and M. Yus, Eur. J. Org. Chem., 2007, 1599–1605 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR and HRMS spectra of all compounds. See DOI: 10.1039/c2ra20213a |
| ‡ CDRI communication number: 8201 |
| This journal is © The Royal Society of Chemistry 2012 |