Highly regioselective C–H bond functionalization: palladium-catalyzed arylation of substituted imidazo[1,2-a]pyridine with aryl chlorides†
Hua
Cao
*,
Yuanguang
Lin
,
Haiying
Zhan
,
Zuodong
Du
,
Xiulian
Lin
,
Qi-Mei
Liang
and
Hong
Zhang
School of Chemistry and Chemical Engineering, Guangdong Pharmaceutical University, Guangzhou 510006, P. R. of China. E-mail: hua.cao@mail.scut.edu.cn; Fax: (+)86(760)88207939
First published on 15th May 2012
Abstract
We have developed an efficient Pd-catalyzed regioselective arylation of substituted imidazo[1,2-a]pyridines with aryl chlorides, which is rarely reported. This methodology has been successfully applied to the synthesis of a variety of substituted imidazo[1,2-a]pyridine core π systems which exhibit a wide range of biological activities in many drugs.
Bicyclic heteroarenes are one of the most important classes of organic compounds because of the abundance of the bicyclic heterocycle structural motif in natural products, and many pharmaceutically relevant and biologically active compounds.1 Thus, the development of efficient transition metal-catalyzed cross-coupling reactions to construct bicyclic heteroarenes is one of the current concerns in chemical transformations that drives increasing interest.2
Organic chemists have sought to develop new and more efficient aryl–aryl bond-forming methods during the past decade and significant advances to this field have been successfully supplanted by transition-metal-catalyzed direct C–H activation/functionalization.3 Among the metal catalysts, the palladium-,4 copper-,5 rhodium-,6 ruthenium-,7 nickel-catalyzed8 C–H bond functionalization of heterocycles are the most important and efficient methods to form complex bicyclic heterocycles molecules. However, aryl bromides or iodides were used as substrates in most of these arylations, and there have only been rare examples for the arylation using less expensive aryl chlorides. Recently, elegant arylations of aryl chlorides as a substrate were reported in such reactions using palladium in the presence of a phosphine ligand with good yields.9 Thus, we hope to develop a convenient arylation reaction of substituted imidazo[1,2-a]pyridine with aryl chlorides via Pd-catalyzed highly regioselective C–H bond functionalization.
The imidazo[1,2-a]pyridine moiety is present in a large number of heterocyclic compounds10 and has been found as a key structural unit in many important pharmaceuticals such as zolpidem (hypnotic), alpidem (anxiolytic), and zolimidine (antiulcer).11 Due to their wide range of biological activities,12 it is still essential to design and develop new methods for the synthesis of these biological compounds. Herein our interest is to develop a highly regioselective direct arylation of the imidazo[1,2-a]pyridine moiety, which is rarely reported.
At the outset of our studies, we focused on identifying the catalytic system and reaction conditions for C–H arylation of 2-methylimidazo[1,2-a]pyridine (1a) with chlorobenzene (2a) and the results were summarized in Table 1. A variety of palladium catalysts in conjunction with different ligands, bases, solvents and temperatures were screened. As shown in Table 1, we were pleased to find that treatment of 1a with 2a in the presence of 2.5 mol% of Pd(OAc)2 and 10 mol% PPh3, gave the corresponding product 3aa in 25% yield (Table 1, entry 1). Other palladium catalysts, such as PdCl2, Pd(dba)2, were also tested, and none showed better activities than Pd(OAc)2 (entries 2–3). Next, we investigated the effects of ligands. Although some literatures have reported that ligand-free Pd(OAc)2 is found to efficiently catalyze C–H activation/arylation, it is not true for this reaction (entry 4). When the reaction was carried out in absence of ligand, only a trace amount of arylation product 3aa was detected by GC-MS. Interestingly, among the ligands examined, such as PBu3, BuAd2P, PCy3 and 1,10-phenanthroline (phen), BuAd2P was the best, giving 3aa in 76% yield (entries 5–8). Subsequently, different bases were also employed and the results indicated that Cs2CO3 was found to be a superior base compared with K2CO3, K3PO4, t-BuOK (entries 9–11). In addition, solvent effects were also investigated in the following tests (entries 13–15). It was found that the rate of the arylation reaction was significantly improved and the best yields were obtained in NMP (entry 14). When toluene and DMA were employed, the product 3aa was also obtained in moderate to good yields. Furthermore, the temperature also affected the rate of the reaction. It was indicated that 120 °C was the best condition for this arylation. Therefore, the optimal reaction conditions involved in using Pd(OAc)2 as the catalyst are: BuAd2P as the ligand, NMP as the solvent at 120 °C for 24 h.
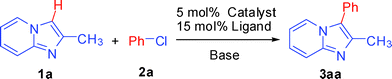
| Entry | Catalyst | Ligand | Base | Solvent | T (°C) | Yield (%)b |
|---|---|---|---|---|---|---|
| a Reaction conditions:1 (0.5 mmol), aryl chlorides (0.7 mmol), Pd-catalyst (2.5 mol%), base (1.5 mmol), ligands (10 mol%), solvent (2 mL), rt–140 °C, 24 h. b GC yields. | ||||||
| 1 | Pd(OAc)2 | PPh3 | K2CO3 | DMF | 100 | 25 |
| 2 | PdCl2 | PPh3 | K2CO3 | DMF | 100 | 24 |
| 3 | Pd(dba)2 | PPh3 | K2CO3 | DMF | 100 | 29 |
| 4 | Pd(OAc)2 | — | K2CO3 | DMF | 100 | trace |
| 5 | Pd(OAc)2 | PBu3 | K2CO3 | DMF | 100 | 30 |
| 6 | Pd(OAc)2 | BuAd2P | K2CO3 | DMF | 100 | 76 |
| 7 | Pd(OAc)2 | PCy3 | K2CO3 | DMF | 100 | 53 |
| 8 | Pd(OAc)2 | Phen | K2CO3 | DMF | 100 | 8 |
| 9 | Pd(OAc)2 | BuAd2P | Cs2CO3 | DMF | 100 | 78 |
| 10 | Pd(OAc)2 | BuAd2P | K3PO4 | DMF | 100 | 41 |
| 11 | Pd(OAc)2 | BuAd2P | t-BuOK | DMF | 100 | 32 |
| 12 | Pd(OAc)2 | BuAd2P | CsOAc | DMF | 100 | 75 |
| 13 | Pd(OAc)2 | BuAd2P | Cs2CO3 | Toluene | 100 | 64 |
| 14 | Pd(OAc)2 | BuAd2P | Cs2CO3 | NMP | 100 | 82 |
| 15 | Pd(OAc)2 | BuAd2P | Cs2CO3 | DMA | 100 | 80 |
| 16 | Pd(OAc)2 | BuAd2P | Cs2CO3 | NMP | 120 | 88 |
| 17 | Pd(OAc)2 | BuAd2P | Cs2CO3 | NMP | 140 | 85 |
| 18 | Pd(OAc)2 | BuAd2P | Cs2CO3 | NMP | 80 | 56 |
| 19 | Pd(OAc)2 | BuAd2P | Cs2CO3 | NMP | rt | — |
Under optimized reaction conditions, the scope with respect to substituted imidazo[1,2-a]pyridine 1 and aryl chlorides 2 was investigated and the results were presented in Table 2. As shown in Table 2, the optimized reaction conditions could be applied to a wide variety of substrates. As expected, a series of functional groups on the phenyl ring of aryl chlorides, such as CH3, C2H5, F, Cl, CN and C(CH3)3, were compatible under this transformation, and the corresponding products 3aa–3al were isolated in moderate to good yields. However, the corresponding products (3am, 3an, 3ao) were not detected when aryl chlorides with strong electron-withdrawing and electron-rich groups were used in the reactions, such as 2-chlorothiophene, 1-chloro-4-nitrobenzene and 1-chloro-4-methoxybenzene. From the results in Table 2, it was clearly indicated that this protocol was general and applicable for the arylation of electron-rich and electron-withdrawing groups on the phenyl of aryl chlorides. Meanwhile, functional groups including electron-rich and electron-withdrawing groups in para and meta positions to the chloride were also well tolerated in arylation reactions. Notably, the sterically hindered aryl chloride also proceeded smoothly, giving the desired arylation products 3af, 3ai in 72% and 83% yields respectively. Unfortunately, the corresponding products were not detected, when the reaction carried was out in optimal conditions and 1-chloro-4-nitrobenzene was used as substrate. After a broad scope of electrophiles was established, we were particularly interested in the effect of the group at the imidazo[1,2-a]pyridine for the arylation process. Substituted imidazo[1,2-a]pyridines, such as 6-chloro-2-methylimidazo[1,2-a]pyridine (1b), 2-(trifluoromethyl)imidazo [1,2-a]pyridine (1c), ethyl 5-methylimidazo[1,2-a]pyridine-2-carboxylate (1d), were also employed and coupled with aryl chlorides to afford the corresponding arylation products in good yield. It was worth noting that the electron-withdrawing groups, such as CF3, CO2Et, at the C-2 of imidazo[1,2-a]pyridine also well worked in the transformation.
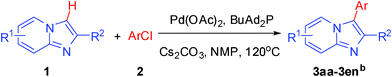

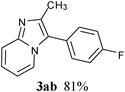










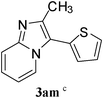


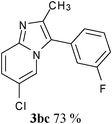



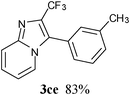


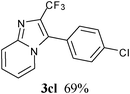





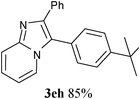

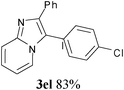


In an endeavour to expand the scope of the methodology, this catalytic system was applied to 2-phenylimidazo[1,2-a]pyridine. To our delight, 2-phenylimidazo[1,2-a]pyridine 1e and aryl chlorides exclusively afforded the corresponding products in moderate to excellent yields (80–85%) via regioselective C–H bond functionalization under the optimized reaction conditions. Significant steric and electronic effects of the substituents on the reactivity were observed. The results revealed that an electron-withdrawing (3ec, 3ed) and electron-rich group (3ea, 3eh, 3ei, 3em) in the aryl chlorides favored the coupling reactions. It also indicated that steric hindrance (3ei) in the aryl chlorides had no influence on the arylation reaction. This process represented the general method for C-3 regioselective arylation of 2-phenylimidazo[1,2-a]pyridine to form a variety of functionalized 2-phenylimidazo[1,2-a]pyridine core π systems. The highly regioselective results observed in Table 2 are possibly because the Ph– at the 2-postition is beneficial to make intermediate B (Scheme 1) stable.
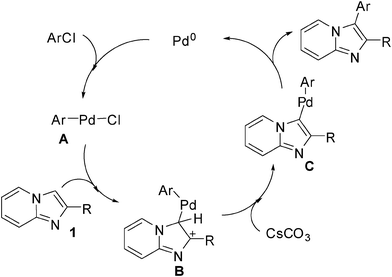 | ||
| Scheme 1 Possible mechanism. | ||
A plausible mechanism of the direct arylation is described in Scheme 1. An analogous mechanism has been proposed by Miura,13 Daugulis9a and Gevorgyan14 for the arylation of heterocycles with aryl halides. We believe that it involves an electrophilic attack by intermediate A on 1 to generate the intermediate B. The deprotonation of intermediate B with Cs2CO3 has been taken place to give intermediate C, which then undergoes reductive elimination to form the corresponding arylation products and release the Pd catalyst.
In summary, a facile and robust method has been developed for the regioselective arylation at the C-3 position of substituted imidazo[1,2-a]pyridine with aryl chlorides by Pd(II)-catalysis. This protocol easily forms a π-conjugated imidazo[1,2-a]pyridine derivative which is a useful structural motif in both academic and industrial research. Further studies for the construction of other heterocyclic ring systems as well as development of novel drug templates using this method are underway.
References
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS; (c) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS; (d) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS.
- (a) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 3850–3850 CrossRef CAS; (b) L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211–229 CrossRef CAS; (c) M. Miura and M. Nomura, Top. Curr. Chem., 2002, 219, 211–241 CrossRef CAS; (d) O. Daugulis, H. Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS; (e) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC.
- (a) L. C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253–1264 RSC; (b) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS; (c) O. Daugulis, V. G. Zaitsev, D. Shabashov, Q. N. Pham and A. Lazareva, Synlett, 2006, 3382–3388 CrossRef CAS; (d) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS; (e) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS.
- (a) E. T. Nadres, A. Lazareva and O. Daugulis, J. Org. Chem., 2011, 76, 471–483 CrossRef CAS; (b) H. A. Ioannidou and P. A. Koutentis, Org. Lett., 2011, 13, 1510–1513 CrossRef CAS; (c) S. Kirchberg, S. Tani, K. Ueda, J. Yamaguchi, A. Studer and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 2387–2391 CAS; (d) K. Ueda, S. Yanagisawa, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 8946–8949 CrossRef CAS; (e) S. Tamba, Y. Okubo, S. Tanaka, D. Monguchi and A. Mori, J. Org. Chem., 2010, 75, 6998–7001 CrossRef CAS; (f) C. L.C. Caron and K. Fagnou, Org. Lett., 2008, 10, 4533–4536 CrossRef; (g) K. Kobayashi, A. Sugie, M. Takahashi, K. Masui and A. Mori, Org. Lett., 2005, 7, 5083–5085 CrossRef CAS; (h) O. Daugulis and V. G. Zaitsev, Angew. Chem., Int. Ed., 2005, 44, 4046–4048 CrossRef CAS; (i) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050–8057 CrossRef CAS; (j) J. P. Leclerc and K. Fagnou, Angew. Chem., Int. Ed., 2006, 45, 7781–7786 CrossRef CAS; (k) A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, J. Am. Chem. Soc., 2003, 125, 1700–1701 CrossRef CAS; (l) M. Zhang, S. Zhang, M. Liu and J. Cheng, Chem. Commun., 2011, 47, 11522–11524 RSC; (m) B. B. Touré, B. S. Lane and D. Sames, Org. Lett., 2006, 8, 1979–1982 CrossRef; (n) H. Guo, W. Rao, H. Niu, L. Jiang, Y. Zhang and G. Qu, RSC Adv., 2011, 1, 961–963 RSC.
- (a) H. Q. Do and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 13577–13586 CrossRef CAS; (b) I. Popov, S. Lindeman and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 9286–9289 CrossRef CAS; (c) G. Huang, H. Sun, X. Qiu, C. Jin, C. Lin, Y. Shen, J. Jiang and L. Wang, Org. Lett., 2011, 13, 5224–5227 CrossRef CAS; (d) T. Truong and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 4243–4245 CrossRef CAS; (e) T. Kawano, T. Yoshizumi, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 3072–3075 CrossRef CAS.
- (a) A. M. Berman, J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 14926–14927 CrossRef CAS; (b) J. C. Lewis, A. M. Berman, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 2493–2500 CrossRef CAS; (c) J. C. Lewis, J. Y. Wu, R. G. Bergman and J. A. Ellman, Angew. Chem., Int. Ed., 2006, 45, 1589–1591 CrossRef CAS; (d) S. Proch and R. Kempe, Angew. Chem., Int. Ed., 2007, 46, 3135–3138 CrossRef CAS.
- (a) W. Li, P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2315–2319 RSC; (b) B. Stefane, J. Fabris and F. Požzgan, Eur. J. Org. Chem., 2011, 3474–3481 CrossRef CAS; (c) P. Arockiam, V. Poirier, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2009, 11, 1871–1875 RSC; (d) L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299–2302 CrossRef CAS; (e) I. Özdemir, S. Demir, B. Cetinkaya, C. Gourlaouen, F. Maseras, C. Bruneau and P. H. Dixneuf, J. Am. Chem. Soc., 2008, 130, 1156–1157 CrossRef; (f) L. Ackermann, A. Althammer and R. Born, Angew. Chem., Int. Ed., 2006, 45, 2619–2622 CrossRef CAS; (g) L. Ackermann, Org. Lett., 2005, 7, 3123–3125 CrossRef CAS.
- (a) G. R. Qu, P. Y. Xin, H. Y. Niu, D. C. Wang, R. F. Dinga and H. M. Guo, Chem. Commun., 2011, 47, 11140–11142 RSC; (b) H. Hachiya, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2010, 49, 2202–2205 CrossRef CAS; (c) H. Hachiya, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 1737–1740 CrossRef CAS.
- (a) H. A. Chiong and O. Daugulis, Org. Lett., 2007, 9, 1449–1451 CrossRef CAS; (b) P. V. Kumar, W. S. Lin, J. S. Shen, D. Nandi and H. M. Lee, Organometallics, 2011, 30, 5160–5169 CrossRef CAS.
- (a) A. Linton, P. Kang, M. Ornelas, S. Kephart, Q. Hu, M. Pairish, Y. Jiang and C. Guo, J. Med. Chem., 2011, 54, 7705–7712 CrossRef CAS; (b) S. Husinec, R. Markovic, M. Petkovic, V. Nasufovic and V. Savic, Org. Lett., 2011, 13, 2286–2289 CrossRef CAS; (c) S. M. Hanson, E. V. Morlock, K. A. Satyshur and C. Czajkowski, J. Med. Chem., 2008, 51, 7243–7252 CrossRef CAS; (d) G. Trapani, M. Franco, A. Latrofa, L. Ricciardi, A. Carotti, M. Serra, E. Sanna, G. Biggio and G. Liso, J. Med. Chem., 1999, 42, 3934–3941 CrossRef CAS.
- (a) M. Z. Mintzer, J. M. Frey and R. R. Griffiths, Behav. Pharmacol., 1998, 9, 545–559 CrossRef CAS; (b) N. Chernyak and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2743–2746 CrossRef CAS; (c) R. Chermat, Y. Minaire, M. C. Chesneau and P. Simon, Therapie, 32, 643–648 CAS.
- (a) K. C. Rupert, J. R. Henry, J. H. Dodd, S. A. Wadsworth, D. E. Cavender, G. C. Olini, B. Fahmy and J. Siekierka, Bioorg. Med. Chem. Lett., 2003, 13, 347–350 CrossRef CAS; (b) Y. Abe, H. Kayakiri, S. Satoh, T. Inoue, Y. Sawada, K. Imai, N. Inamura, M. Asano, C. Hatori, A. Katayama, T. Oku and H. Tanaka, J. Med. Chem., 1998, 41, 564–578 CrossRef CAS; (c) C. Hamdouchi, B. Zhong, J. Mendoza, E. Collins, C. Jaramillo, J. E. De Diego, D. Robertson, C. D. Spencer, B. D. Anderson, S. A. Watkins, F. Zhanga and H. B. Brooks, Bioorg. Med. Chem. Lett., 2005, 15, 1943–1947 CrossRef CAS.
- S. Pivsa-Art, T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 467–473 CrossRef CAS.
- C. H. Park, V. Ryabova, I. V. Seregin, A. W. Sromek and V. Gevorgyan, Org. Lett., 2004, 6, 1159–1162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20366a |
| This journal is © The Royal Society of Chemistry 2012 |