Ruthenium(II) carbonyl complexes containing benzhydrazone ligands: synthesis, structure and facile one-pot conversion of aldehydes to amides†
Rupesh Narayana
Prabhu
and
Rengan
Ramesh
*
School of Chemistry, Bharathidasan University, Tiruchirappalli 620 024, Tamil Nadu, India. E-mail: ramesh_bdu@yahoo.com; Tel: +91-431-2407053; Fax: +91-431-2407045/2407020
First published on 5th March 2012
Abstract
Convenient synthesis of a series of eight octahedral ruthenium(II) carbonyl complexes having the general molecular formula [Ru(L)(CO)Cl(AsPh3)2] (where HL = thiophene aldehyde benzhydrazone ligand) has been described. The substituted benzhydrazone ligands behave as monoanionic bidentate N and O donors (L) and coordinate to the ruthenium(II) ion via the azomethine nitrogen and deprotonated amide oxygen. The compositions of the complexes have been established by elemental analysis, spectral methods (FT-IR, UV-Vis, 1H NMR) and X-ray crystallography. The crystal structure of two of the complexes, viz., [Ru(L1)(CO)Cl(AsPh3)2] (1) and [Ru(L5)(CO)Cl(AsPh3)2] (5) have been solved by single crystal X-ray crystallography and it indicates the presence of a distorted octahedral geometry in these complexes. The complexes were demonstrated as efficient catalysts for the one-pot conversion of various aldehydes to their corresponding primary amides using NH2OH·HCl and NaHCO3. The effects of solvent, base, reaction temperature and catalyst loading on the catalytic activity of the most active ruthenium(II) carbonyl benzhydrazone complex were also investigated.
Introduction
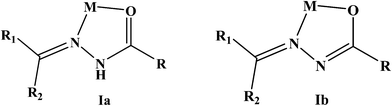
There has been continuous research activities towards the design of new types of ruthenium(II) complexes containing diimine ligands as well as ligands containing oxygen and nitrogen donor atoms. This is primarily due to their kinetic inertness (low-spin d6), energy transfer properties, multi-electron transfer properties and their ability to have a wide range of oxidation states which in turn depend on the nature of the ligands bound to the metal.1 The activity of the ruthenium complexes containing various multidentate ligands in catalytic applications has also been well documented.2 In particular, triphenylphosphine and triphenylarsine ligands are well known to stabilize transition metals in low as well as high oxidation states and also increase the solubility of the complexes in less polar solvents.3Aroyl hydrazones are versatile ligands exhibiting amide-imidol tautomerism and display interesting coordination modes in metal complexes.4 Depending on the acidity, the reaction conditions and the nature of the metal ion, these ligands coordinate to the metal ion (Scheme 1), either in the neutral amide form (Ia) or in the monoanionic imidolate form (Ib), as bidentate N, O donor ligands forming five-membered chelate rings with the metal.5
 | ||
| Scheme 1 Coordination modes of acid hydrazone ligands. | ||
Several metal complexes containing acid hydrazone ligands have been reported to have potential applications as catalysts, luminescent probes and potential iron chelators or anticancer agents.6 Compared with the considerable work which has been published relating to the use of acid hydrazones as ligands for first-row transition metal and lanthanide metal complexes, only a few have been reported on ruthenium(II) complexes. Ru(II) bis- and mixed-ligand complexes with aroyl (picolinylidene)hydrazine ligands were reported where the energy gap between the metal-dπ and ligand-π* levels remains unaffected by the change of polar effect on the substituent on the aroyl fragment of the ligand.7 Further, Ru(II) complexes containing heterocyclic hydrazone ligands have been structurally characterized8 and investigated for cancer chemotherapy, antibacterial activity and DNA binding.9
The development of efficient methods for the synthesis of amides is very important because of their usefulness in a wide variety of applications in academia and industry, especially as intermediates in organic synthesis, raw materials for engineering plastics, detergents, lubricants and pharmaceuticals.10 Amides are commonly prepared from the stoichiometric reaction of amines with acyl chlorides, acid anhydrides and esters or involve the use of stoichiometric coupling reagents such as carbodiimides11 or by the metal-mediated hydrolysis of organonitriles.12 However, the toxicity and waste formation involved in these methods has made the atom-economical synthesis of amides a high priority, especially in the pharmaceutical industry.
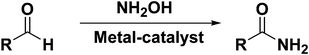
Transition metal catalyzed one-pot conversion of aldehydes to amides in presence of NH2OH (Scheme 2) has been recently investigated. Rh(OH)x supported over alumina was reported as an effective heterogeneous catalyst for the one-pot synthesis of primary amides from various aldehydes and NH2OH·H2SO4 in water at elevated temperatures with the formation of unwanted nitriles, aldoximes and carboxylic acids as by-products.13 Simple inexpensive zinc salts were found to be good homogeneous catalysts for the conversion of aldehydes into primary amides and nitriles using NH2OH·HCl and NaHCO3 in moderate conversions under high catalyst loading.14 Ru(II) complexes, viz., [Ru(terpy)(PPh3)2Cl2]15 and [Ru(DMSO)4Cl2]16 were also used as homogeneous catalysts for this transformation using NH2OH·HCl and NaHCO3. Recently FeCl3 was also employed as a catalyst using Cs2CO3 as a base in aqueous medium for the transformation of aldehydes into primary amides.17
 | ||
| Scheme 2 Metal catalyzed conversion of aldehydes to amides. | ||
In continuation of our research on the synthesis, characterization and catalytic applications of ruthenium, rhodium and palladium complexes18 and in view of the interesting coordination modes of aroylhydrazone ligands, we herein describe new Ru(II) carbonyl complexes with substituted thiophene aldehyde benzhydrazone ligands incorporated with chloride and triphenylarsine as ancillary ligands. All the complexes have been characterized by analytical and spectral methods. The structures of two of the complexes have been probed with the help of single crystal X-ray diffraction analysis. The application of these complexes as homogeneous catalyst for the one pot conversion of aldehydes to corresponding primary amides using NH2OH·HCl was also investigated.
Results and discussion
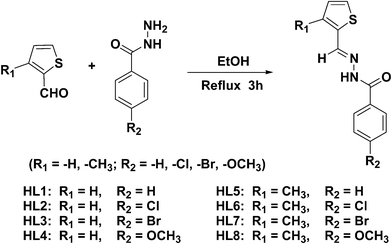
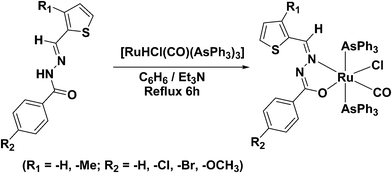
The benzhydrazone ligand derivatives were conveniently prepared in an excellent yield by the condensation of substituted thiophene aldehydes with substituted benzhydrazides in an equimolar ratio (Scheme 3). These ligands were allowed to react with the ruthenium(II) precursor, [RuHCl(CO)(AsPh3)3] in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in the presence of triethylamine as the base and the new complexes of the of the general formula, [Ru(L)(CO)Cl(AsPh3)2] (Scheme 4) were obtained in reasonable yields. The addition of triethylamine to the reaction mixture was used to abstract a proton from the imidol oxygen and to facilitate the coordination of the imidolate oxygen to the ruthenium(II) ion. It has been observed that the ligands behave as monoanionic bidentates, replacing one hydride and one triphenylarsine from the ruthenium(II) precursor; the oxidation state of ruthenium remain unchanged during the formation of the complex. All the complexes are yellow in colour, air stable in both the solid and the liquid states at room temperature and are non-hygroscopic in nature. The synthesized ruthenium(II) complexes are soluble in common solvents such as chloroform, dichloromethane, acetonitrile, dimethyl formamide, dimethyl sulphoxide etc., producing intense coloured solutions. The analytical data of all the ruthenium(II) complexes are in good agreement with the molecular structures proposed.
:1 molar ratio in the presence of triethylamine as the base and the new complexes of the of the general formula, [Ru(L)(CO)Cl(AsPh3)2] (Scheme 4) were obtained in reasonable yields. The addition of triethylamine to the reaction mixture was used to abstract a proton from the imidol oxygen and to facilitate the coordination of the imidolate oxygen to the ruthenium(II) ion. It has been observed that the ligands behave as monoanionic bidentates, replacing one hydride and one triphenylarsine from the ruthenium(II) precursor; the oxidation state of ruthenium remain unchanged during the formation of the complex. All the complexes are yellow in colour, air stable in both the solid and the liquid states at room temperature and are non-hygroscopic in nature. The synthesized ruthenium(II) complexes are soluble in common solvents such as chloroform, dichloromethane, acetonitrile, dimethyl formamide, dimethyl sulphoxide etc., producing intense coloured solutions. The analytical data of all the ruthenium(II) complexes are in good agreement with the molecular structures proposed.
 | ||
| Scheme 3 Preparation of benzhydrazone ligands. | ||
 | ||
| Scheme 4 Synthesis of Ru(II) carbonyl benzhydrazone complexes. | ||
The IR spectra of the free ligands showed a medium to strong band in the region 3191–3280 cm−1 which is characteristic of the N–H functional group. The free ligands also display νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and νCO absorptions in the region 1632–1649 cm−1. The bands due to νN–H and νCO stretching vibrations are not observed in the complexes indicating that the ligands undergo tautomerization and subsequent coordination of the imidolate form during complexation. This is further supported by the appearance of new bands in the range 1256–1292 and 1590–1605 cm−1, which may be attributed to the C–O and CN–NC fragments respectively of the coordinated ligand.19 All the complexes display a medium to strong band in the region 1935–1946 cm−1, which is attributed to the terminally coordinated carbonyl group and is observed at a slightly higher frequency than in the precursor complexes. In addition, other characteristic bands due to ruthenium bound triphenylarsine are also present in the region 1434–1484 cm−1 in the spectra of all the complexes.20 The IR spectra of all the complexes therefore confirms the coordination mode of the benzhydrazone ligand to the ruthenium(II) ion via the azomethine nitrogen and the imidolate oxygen along with the presence of triphenylarsine groups.
N and νCO absorptions in the region 1632–1649 cm−1. The bands due to νN–H and νCO stretching vibrations are not observed in the complexes indicating that the ligands undergo tautomerization and subsequent coordination of the imidolate form during complexation. This is further supported by the appearance of new bands in the range 1256–1292 and 1590–1605 cm−1, which may be attributed to the C–O and CN–NC fragments respectively of the coordinated ligand.19 All the complexes display a medium to strong band in the region 1935–1946 cm−1, which is attributed to the terminally coordinated carbonyl group and is observed at a slightly higher frequency than in the precursor complexes. In addition, other characteristic bands due to ruthenium bound triphenylarsine are also present in the region 1434–1484 cm−1 in the spectra of all the complexes.20 The IR spectra of all the complexes therefore confirms the coordination mode of the benzhydrazone ligand to the ruthenium(II) ion via the azomethine nitrogen and the imidolate oxygen along with the presence of triphenylarsine groups.
The 1H NMR spectra of all the complexes were recorded in CDCl3 to confirm the binding of the benzhydrazone ligands to the ruthenium(II) ion. Multiplets observed in the region δ 6.6–8.0 ppm in all the complexes have been assigned to the aromatic protons of triphenylarsine and benzhydrazone ligands. The signal due to the azomethine proton appears as a sharp singlet in the region δ 9.9–10.0 ppm. The positions of the azomethine signal in the complexes are downfield in comparison with those of the free ligands, suggesting deshielding of the azomethine proton due to its coordination to ruthenium.21 A sharp singlet that appeared for the –NH proton of the free ligand in the region δ 11.6–11.9 ppm is absent in all the complexes, further supporting enolisation and coordination of the imidolate oxygen to the ruthenium(II) ion. For the complexes (5), (6), (7) and (8) additional methyl signals of the thiophene ring are observed as a singlet at δ 2.5 ppm, whereas for complexes (4) and (8) the methoxy signals of the benzhydrazone ring are observed as a singlet at δ 3.6–3.8 ppm. The 1H NMR spectra of all the complexes further supports the coordination mode of the benzhydrazone ligand to the ruthenium(II) ion via the azomethine nitrogen and the imidolate oxygen along with the presence of two triphenylarsine groups. The 1H NMR spectra for all the complexes are shown in Fig. S1–S8 (ESI†).
The absorption spectra of all the complexes in acetonitrile at room temperature showed three bands in the region 247–404 nm. The high intensity bands in the 247–336 nm region were assigned to ligand-centered (LC) transitions and have been designated as π–π* and n–π* transitions. In all the complexes the lowest energy bands observed in the region 394–404 nm were attributed to the charge transfer due to metal to ligand transitions with possible contributions from ligand centered transitions. The fact that there is essentially no variation in the energy of the MLCT band suggests that the energy gap between the metal-dπ and the ligand π* levels remains constant despite the variation of the substituent on the aroyl/thiophene fragment of the ligand.7 The pattern of the electronic spectra of all the complexes indicated the presence of an octahedral environment around the ruthenium(II) ion is similar to that of other octahedral ruthenium(II) complexes.22
The molecular structure of two of the complexes, [Ru(L1)(CO)Cl(AsPh3)2] (1) and [Ru(L5)(CO)Cl(AsPh3)2] (5) have been determined by single crystal X-ray diffraction to confirm the coordination mode of the benzhydrazone ligand in the complexes and the stereochemistry of the complexes. The summary of the data collected and the refinement parameters are given in Table 1 whereas selected bond lengths and bond angles are given in Table 2 and 3. The ORTEP view of complex (1) is shown in Fig. 1. The complex crystallizes in the P21/c space group. The benzhydrazone ligand coordinates in a bidentate manner to the ruthenium(II) ion via the azomethine nitrogen and the deprotonated amide oxygen in the benzhydrazone fragment, forming one five-membered chelate ring. One carbonyl group (trans to the azomethine nitrogen) and one chloride ion also coordinate to the Ru(II) ion to form a CNOCl square-plane and the arsine atoms of the two triphenylarsine ligands occupy the two axial sites. Ruthenium is therefore sitting in a CNOClAs2 coordination environment, which is distorted octahedral in nature as reflected in all the bond parameters around ruthenium. The bite angles around the Ru(II) ion are C(49)–Ru(1)–O(1) = 93.8(4)°, N(1)–Ru(1)–O(1) = 76.2(3)°, C(49)–Ru(1)–Cl(1) = 96.8(3)° and N(1)–Ru(1)–Cl(1) = 93.3(3)°, and bond lengths of 2.395(3) Å Ru(1)–Cl(1), 2.073(6) Å Ru(1)–O(1), 2.070(7) Å Ru(1)–N(1) and 1.944(10) Å Ru(1)–C(49). Further the ORTEP view of complex (5) is shown in Fig. 2. It was observed that complex (5) also adopts a similar geometry as in complex (1) with slight changes in bond angles and bond lengths. The bond lengths and bond angles are in good agreement with reported data on related ruthenium(II) complexes.23 As all the complexes display similar spectral properties, the other six complexes are assumed to have a similar structure to that of complexes (1) and (5).
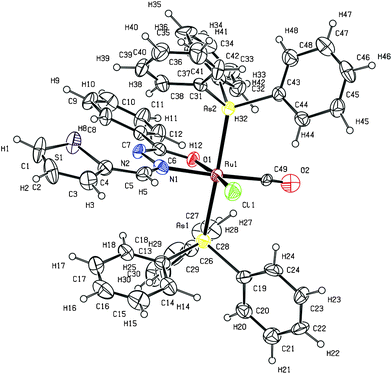 | ||
| Fig. 1 ORTEP diagram of the complex (1). Displacement ellipsoids are drawn at the 30% probability level. | ||
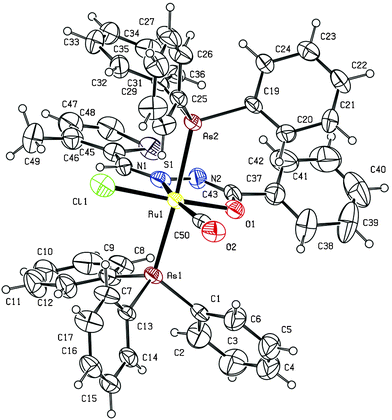 | ||
| Fig. 2 ORTEP diagram of the complex (5). Displacement ellipsoids are drawn at the 30% probability level. | ||
| Complex (1) | Complex (5) | |
|---|---|---|
| Empirical formula | C49H39As2Cl1N2O2RuS | C50H41As2Cl1N2O2RuS |
| Formula weight | 1006.25 | 1020.27 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | C2/c |
| a (Å) | 16.860(5) | 41.1816(10) |
| b (Å) | 11.836(5) | 11.3351(3) |
| c (Å) | 23.186(5) | 40.5037(11) |
| α (°) | 90 | 90 |
| β (°) | 109.358(5) | 108.586(3) |
| γ (°) | 90 | 90 |
| Volume | 4365(2) Å3 | 17921.0(8) Å3 |
| Z, Calculated density | 4, 1.531 Mg m−3 | 2, 0.189 Mg m−3 |
| Absorption coefficient | 2.014 mm−1 | 0.245 mm−1 |
| F(000) | 506 | 1028 |
| Theta range | 1.28 to 19.15 deg. | 1.04 to 20.71 deg. |
| Limiting indices | −15 ≤ h ≤ 15,−10 ≤ k ≤ 10, −21 ≤ l ≤ 21 | −40 ≤ h ≤ 40, −11 ≤ k ≤ 11, −40 ≤ l ≤ 40 |
| Reflections collected/unique | 39499/3588 [R(int) = 0.1151] |
78532/9250 [R(int) = 0.0652] |
| Completeness to θmax | 100.0% | 100.0% |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3588/0/659 | 9250/0/1063 |
| Goodness-of-fit on F2 | 1.084 | 1.077 |
| Final R indices [I > 2σ(I)] | R1 = 0.0346, wR2 = 0.0866 | R1 = 0.0436, wR2 = 0.0950 |
| R indices (all data) | R1 = 0.0659, wR2 = 0.0649 | R1 = 0.0837, wR2 = 0.1159 |
| Largest diff. peak & hole | 0.402 and −0.338 e.A−3 | 0.467 and −0.518 e.A−3 |
| Bond length (Å) | Bond angle (°) | ||
|---|---|---|---|
| a ESD in parenthesis. | |||
| Ru1–As1 | 2.4626(13) | C49–Ru1–N1 | 169.9(4) |
| Ru1–As2 | 2.4739(13) | C49–Ru1–O1 | 93.8(4) |
| Ru1–C49 | 1.944(10) | N1–Ru1–O1 | 76.2(3) |
| Ru1–Cl1 | 2.395(3) | C49–Ru1–Cl1 | 96.8(3) |
| Ru1–N1 | 2.070(7) | N1–Ru1–Cl1 | 93.3(3) |
| Ru1–O1 | 2.073(6) | O1–Ru1–Cl1 | 169.40(19) |
| As1–C13 | 1.912(9) | C49–Ru1–As1 | 90.8(3) |
| As1–C19 | 1.963(10) | N1–Ru1–As1 | 89.94(19) |
| As1–C26 | 1.935(9) | O1–Ru1–As1 | 89.23(16) |
| As2–C31 | 1.952(10) | Cl1–Ru1–As1 | 91.76(7) |
| As2–C37 | 1.930(9) | C49–Ru1–As2 | 89.7(3) |
| As2–C43 | 1.936(9) | N1–Ru1–As2 | 89.07(19) |
| S1–C1 | 1.709(13) | O1–Ru1–As2 | 87.98(16) |
| S1–C4 | 1.713(10) | Cl1–Ru1–As2 | 90.92(7) |
| O1–C6 | 1.271(10) | As1–Ru1–As2 | 177.19(5) |
| C6–C7 | 1.522(13) | N2–N1–Ru1 | 115.7(6) |
| O2–C49 | 0.924(8) | C6–O1–Ru1 | 111.4(6) |
| N2–N1 | 1.409(10) | C5–N1–N2 | 115.0(9) |
| N2–C6 | 1.316(10) | C5–N1–Ru1 | 129.4(8) |
| C6–N2–N1 | 108.5(7) | ||
| O1–C6–N2 | 127.4(9) | ||
| N2–C6–C7 | 116.5(10) | ||
| O1–C6–C7 | 116.1(9) | ||
| Bond length (Å) | Bond angle (°) | ||
|---|---|---|---|
| a ESD in parenthesis. | |||
| Ru1–As1 | 2.4632(10) | C50–Ru1–N1 | 168.0(5) |
| Ru1–As2 | 2.4810(10) | C50–Ru1–O1 | 90.1(5) |
| Ru1–C50 | 1.981(11) | C50–Ru1–Cl1 | 98.3(4) |
| Ru1–Cl1 | 2.409(3) | O1–Ru1–N1 | 77.9(3) |
| Ru1–N1 | 2.082(8) | O1–Ru1–Cl1 | 171.6(2) |
| Ru1–O1 | 2.062(6) | N1–Ru1–Cl1 | 93.7(3) |
| As1–C1 | 1.927(9) | C50–Ru1–As1 | 91.8(4) |
| As1–C7 | 1.951(9) | N1–Ru1–As1 | 89.02(18) |
| As1–C13 | 1.961(9) | O1–Ru1–As1 | 89.48(15) |
| As2–C19 | 1.937(8) | Cl1–Ru1–As1 | 90.82(7) |
| As2–C25 | 1.938(9) | C50–Ru1–As2 | 93.8(4) |
| As2–C31 | 1.947(9) | N1–Ru1–As2 | 85.80(18) |
| S1–C45 | 1.737(9) | O1–Ru1–As2 | 91.87(15) |
| S1–C48 | 1.689(11) | Cl1–Ru1–As2 | 87.04(7) |
| O1–C43 | 1.280(11) | As1–Ru1–As2 | 174.26(4) |
| N2–C43 | 1.341(12) | O1–C43–N2 | 126.9(9) |
| C50–O2 | 0.849(12) | C43–N2–N1 | 109.5(8) |
| N2–N1 | 1.429(10) | N2–N1–C44 | 115.8(8) |
| N1–C44 | 1.285(10) | N2–N1–Ru1 | 113.9(6) |
| C48–S1–C45 | 90.2(6) | ||
| N2–C43–C37 | 116.9(10) | ||
Catalytic one-pot conversion of aldehydes to amides
The one-pot conversion of various aldehydes to the corresponding amides in the presence of NH2OH·HCl using ruthenium(II) complexes as catalysts was investigated under various conditions. In order to systematically investigate the influence of solvent, base and effect of catalyst loading, a proper model had to be established. The one-pot conversion of 4-nitrobenzaldehyde to 4-nitrobenzamide in the presence of NH2OH·HCl using complex (5) as a catalyst was first examined as a test reaction under various conditions (Table 4). No conversion of 4-nitrobenzaldehyde to 4-nitrobenzamide was observed in the absence of the catalyst or base. The extent of conversion is solvent-dependent and low conversions were observed in toluene, benzene and xylene. Acetonitrile was found to be the solvent of choice at a lower temperature. Since base facilitates the neutralization of NH2OH·HCl, the effect of the base was also investigated in the formation of 4-nitrobenzamide from 4-nitrobenzaldehyde. It has been observed that in acetonitrile solvent, NaHCO3 or KHCO3 gave good isolated yields when compared to a much weaker base like CH3COONa or Et3N. Thus, it was concluded that NaHCO3 as base in acetonitrile solvent at 78 °C is the optimised condition for this conversion.
| Entry | Solvent | Base | Temp. (°C) | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 4-nitrobenzaldehyde (2.98 × 10−4 mol), NH2OH·HCl (2.98 × 10−4 mol), base (2.98 × 10−4 mol), complex (5) (2.98 × 10−6 mol) and solvent (2 mL) were refluxed for a specific amount of time under an N2 atmosphere. b Isolated yield after column chromatography (average of two runs). c Reaction carried out in the absence of catalyst. | |||||
| 1 | Xylene | NaHCO3 | 140 | 36 | 28 |
| 2 | Toluene | NaHCO3 | 110 | 36 | 25 |
| 3 | Benzene | NaHCO3 | 80 | 36 | 20 |
| 4 | CH3CN | NaHCO3 | 78 | 6 | 66 |
| 5 | CH3CN | KHCO3 | 78 | 6 | 64 |
| 6 | CH3CN | CH3COONa | 78 | 6 | < 20 |
| 7 | CH3CN | Et3N | 78 | 6 | < 20 |
| 8 | CH3CN | NaHCO3 | 78 | 36c | — |
| 9 | CH3CN | — | 78 | 36 | — |
In order to optimize the effect of catalyst loading, different catalyst:substrate (C:S) ratios were tested in the one-pot conversion of 4-nitrobenzaldehyde to 4-nitrobenzamide using complex (5) as a catalyst and the results are summarized in Table 5. The reaction proceeds with good isolated yield when the C:S ratio is either 1:100 or 1:200. When changing the C:S ratio to 1:300, 1:400 or 1:500, the reaction still proceeds smoothly accompanied by a drop in the isolated yield. Since the isolated yields are good with appreciable turnover numbers (TON) when the C:S ratio is 1:200, it was concluded that this C:S ratio is the best suitable for the catalytic conversion of aldehydes to amides.
:substrate (C:S) ratio in the one-pot conversion of 4-nitrobenzaldehyde to 4-nitrobenzamide using complex (5)a
| Entry | C:S ratio |
Yieldb (%) | TONc |
|---|---|---|---|
| a Reaction conditions: complex (5) (2.98 × 10−4 mol), 4-nitrobenzaldehyde, NH2OH·HCl and NaHCO3 (equimolar ratio) in MeCN (2 mL) refluxed for 6 h under an N2 atmosphere. b Isolated yield after column chromatography (average of two runs). c TON = Turnover number = ratio of moles of product formed to moles of catalyst used. | |||
| 1 | 1:100 |
66 | 66 |
| 2 | 1:200 |
64 | 128 |
| 3 | 1:300 |
52 | 156 |
| 4 | 1:400 |
38 | 152 |
| 5 | 1:500 |
23 | 115 |
The progress of the formation of 4-nitrobenzamide from 4-nitrobenzaldehyde as a function of time using complex (5) as a catalyst is displayed in Fig. 3. The results indicate that the formation of 4-nitrobenzamide increased initially with the progress of the reaction time, reached a maxim and then remained unchanged. A reasonably good isolated yield for the formation of 4-nitrobenzamide was observed at the optimum reaction time of 10 h (83%), whereas over a period of 18 h the maximum isolated yield (92%) was achieved. Further, the efficiency of all the eight ruthenium(II) complexes towards the one-pot conversion of 4-nitrobenzaldehyde to 4-nitrobenzamide was also investigated (Table 6). It was observed that all the complexes displayed similar catalytic activity suggesting that there is no significant effect on the catalysis despite the change in the substituent on the aroyl/thiophene fragment of the ligand in the complexes.
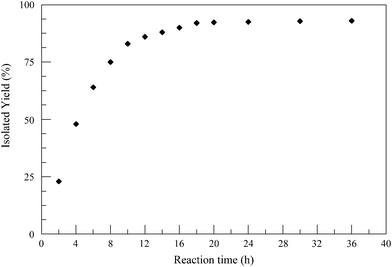 | ||
| Fig. 3 Influence of reaction time on isolated yield. Reaction conditions: complex (5) (2.98 × 10−6 mol), 4-nitrobenzaldehyde (5.96 × 10−4 mol), NH2OH·HCl (5.96 × 10−4 mol) and NaHCO3 (5.96 × 10−4 mol) in MeCN (2 mL) refluxed under an N2 atmosphere; isolated yield after column chromatography (average of two runs). | ||
| Complexes | Yieldb (%) | TONc |
|---|---|---|
| a Reaction conditions: complexes (1)–(8) (2.98 × 10−6 mol), 4-nitrobenzaldehyde (5.96 × 10−4 mol), NH2OH·HCl (5.96 × 10−4 mol) and NaHCO3 (5.96 × 10−4 mol) in MeCN (2 mL) refluxed under an N2 atmosphere for 10 h. b Isolated yield after column chromatography (average of two runs). c TON = Turnover number = ratio of moles of product formed to moles of catalyst used. | ||
| 1 | 85 | 170 |
| 2 | 82 | 164 |
| 3 | 80 | 160 |
| 4 | 81 | 162 |
| 5 | 83 | 166 |
| 6 | 80 | 160 |
| 7 | 81 | 162 |
| 8 | 80 | 160 |
In light of the results obtained, complex (1) shows relatively better catalytic activity among the eight complexes. Hence complex (1) was selected as the model catalyst for the one-pot conversion of various aromatic and heterocyclic aromatic aldehydes to the corresponding primary amides using NH2OH·HCl by refluxing acetonitrile with NaHCO3 as the base, the results are summarized in Table 7. Benzaldehyde with substituents of varying electronic properties were all smoothly converted to the corresponding amides (entries 1–4) in excellent isolated yields and high TON. Electron withdrawing substituents (nitro or bromo) on benzaldehyde gave slightly higher yields (entries 1,2) when compared to that of benzaldehyde (entry 3). Electron donating substituent (methyl or methoxy) on benzaldehyde gave slightly lower yields (entries 4,5) compared with benzaldehyde. The conversion proceeded smoothly even in the presence of heteroatoms such as S and N in the substrates (entries 6–10) and a range of heterocyclic aromatic amides were obtained in good isolated yields. Notably in these reactions other by-products such as organonitriles or carboxylic acids are not observed.
| Entry no. | Product | Yieldb (%) | TONc | TOFd (h) | |||
|---|---|---|---|---|---|---|---|
| 10 h | 18 h | 10 h | 18 h | 10 h | 18 h | ||
| a Reaction conditions: Complex (1) (2.98 × 10−4 mol), aldehyde (5.96 × 10−4 mol), NH2OH·HCl (5.96 × 10−4 mol) and NaHCO3 (5.96 × 10−4 mol) in MeCN (2 mL) refluxed for specific amount of time under an N2 atmosphere. b Isolated yield after column chromatography (average of two runs). c TON = Turnover number = ratio of moles of product formed to moles of catalyst used. d TOF = Turnover frequency = TON (h). | |||||||
| 1 |
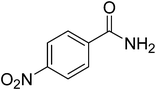
|
85 | 94 | 170 | 188 | 17.0 | 10.4 |
| 2 |
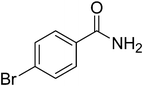
|
83 | 91 | 166 | 182 | 16.6 | 10.1 |
| 3 |
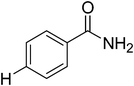
|
81 | 89 | 162 | 178 | 16.2 | 9.9 |
| 4 |
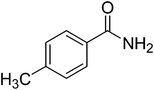
|
75 | 85 | 150 | 170 | 15.0 | 9.4 |
| 5 |
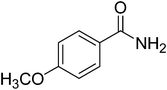
|
70 | 80 | 140 | 160 | 14.0 | 8.9 |
| 6 |
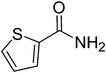
|
76 | 87 | 152 | 174 | 15.2 | 9.7 |
| 7 |
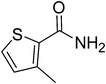
|
73 | 83 | 146 | 166 | 14.6 | 9.2 |
| 8 |
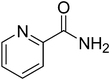
|
72 | 81 | 144 | 162 | 14.2 | 9.0 |
| 9 |
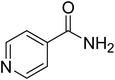
|
73 | 81 | 146 | 162 | 14.6 | 9.0 |
| 10 |
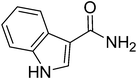
|
76 | 84 | 152 | 168 | 15.2 | 9.3 |
Further, ketones such as acetophenone, 2-acetylthiophene and 2-acetylpyridine were inert to these conditions suggesting that this reaction differs from the standard Beckmann rearrangement. We believe that the catalytic transformation proceeds via the formation of a RuIV(OH)(imine) intermediate as proposed by Crabtree and coworkers;16 however a detailed mechanistic investigation is in progress. In terms of TON/TOF, the new ruthenium(II) carbonyl benzhydrazone complexes are more efficient than [Ru(terpy)(PPh3)2Cl2],15 but less efficient when compared to that of [Ru(DMSO)4Cl2]16.
Conclusion
A series of eight ruthenium(II) carbonyl complexes bearing thiophenealdehyde benzhydrazone ligands of the general formula [Ru(L)(CO)Cl(AsPh3)2] have been synthesized from the reactions of [RuHCl(CO)(AsPh3)3] with substituted thiophene aldehyde benzhydrazone ligands (HL). The characterization of the complexes were accomplished by analytical and spectral (IR, UV-Vis, 1H NMR) methods. X-ray diffraction study of the complexes (1) and (5) confirms the N and O coordination mode of benzhydrazone ligands and reveals the presence of a distorted octahedral geometry around the ruthenium(II) ion. The new complexes act as excellent catalysts for the one-pot conversion of aldehydes to the corresponding amides with good to excellent isolated yields.Experimental
Materials
Commercially available RuCl3·3H2O was used as supplied from Loba Chemie Pvt. Ltd. All the reagents used were chemically pure and AnalaR grade. The solvents were freshly distilled before use following the standard procedures.24 Triphenylarsine, all aldehydes and benzhydrazide derivatives were purchased from Aldrich and were used as received. The ruthenium(II) precursor complex, [RuHCl(CO)(AsPh3)3], was prepared by reported literature method.25Physical measurements and instrumentation
Melting points were recorded in the Boetius micro heating table and are uncorrected. The microanalysis of carbon, hydrogen, nitrogen and sulphur were recorded by a analytical function testing Vario EL III CHNS elemental analyzer at the Sophisticated Test and Instrumentation Centre (STIC), Cochin University, Kochi. The infrared spectra of complexes were recorded in KBr pellets with a Perkin-Elmer 597 spectrophotometer in the range 4000–400 cm−1. The NMR spectra were recorded in DMSO-d6 (for the ligands and amides) and CDCl3 (for the complexes) with a Bruker 400 MHz instrument using TMS as the internal reference. Chemical shifts are given in ppm referenced to solvents. The electronic spectra of the complexes in acetonitrile solution were recorded with a Cary 300 Bio UV-Vis Varian spectrophotometer in the range 800–230 nm.Preparation of benzhydrazone ligands
To a stirred ethanolic solution (10 mL) of 4-substituted benzhydrazide (0.68–1.0 g, 5 mmol), a solution of thiophene-2-aldehyde or 3-methylthiophene-2-aldehyde (0.46–0.53 mL, 56–63 mg, 5 mmol) in ethanol (10 mL) was added dropwise. The reaction mixture was refluxed for 3 h, the solution concentrated to 5 mL and cooled to room temperature. The solid formed was filtered, washed with cold methanol (5 mL) and dried in air.O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.8 (s, 1H, NH), 8.7 (s, 1H, CHN), 7.2–8.0 (m, 8H, aromatic).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.9 (s, 1H, NH), 8.7 (s, 1H, CHN), 7.2–8.0 (m, 7H, aromatic).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.9 (s, 1H, NH), 8.7 (s, 1H, CHN), 7.1–7.9 (m, 7H, aromatic).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.7 (s, 1H, NH), 8.7 (s, 1H, CHN), 7.2–8.0 (m, 7H, aromatic), 3.8 (s, 3H, OCH3).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.7 (s, 1H, NH), 8.7 (s, 1H, CHN), 6.9–8.3 (m, 7H, aromatic), 2.3 (s, 3H, CH3).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.8 (s, 1H, NH), 8.7 (s, 1H, CHN), 6.9–8.0 (m, 6H, aromatic), 2.3 (s, 3H, CH3).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.8 (s, 1H, NH), 8.7 (s, 1H, CHN), 6.9–7.9 (m, 6H, aromatic), 2.3 (s, 3H, CH3).
O) + ν(CN). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 11.6 (s, 1H, NH), 8.7 (s, 1H, CHN), 6.9–7.9 (m, 6H, aromatic), 3.8 (s, 3H, OCH3), 2.3 (s, 3H, CH3).
Synthesis of ruthenium(II) carbonyl complexes containing benzhydrazone ligands
All the reactions were carried out under anhydrous conditions under nitrogen atmosphere and the new ruthenium complexes were prepared by the following general procedure: to a benzene solution (20 mL) of [RuHCl(CO)(AsPh3)3] (100 mg, 0.092 mmol) was added the appropriate benzhydrazone ligand (21.3–29.7 mg, 0.092 mmol) and triethylamine (0.5 mL) as the base. The reaction mixture was refluxed for 6 h and the progress of the reaction was monitored using TLC. At the end of the reaction the solution was concentrated to about 3 mL and light petroleum ether (60–80 °C) was added whereby the solid separated out. The obtained solids were recrystallized from CH2Cl2/petroleum ether and dried under vacuum.N–NC), 1276 m ν(C–O), 1939 s ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 9.9 (s, 1H, CHN), 6.8–8.0 (m, 38H, aromatic). UV-Vis (CH3CN, λmax (nm); ε (dm3mol−1 cm−1): 400(3,660), 335(5,720), 264(14,850).
N–NC), 1263 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 9.9 (s, 1H, CHN), 6.9–8.0 (m, 37H, aromatic). UV-Vis (CH3CN, λmax (nm); ε (dm3 mol−1 cm−1): 404(1,780), 320(10,560), 247(15,130).
N–NC), 1263 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 9.9 (s, 1H, CHN), 6.8–8.0 (m, 37H, aromatic). UV-Vis (CH3CN, λmax (nm); ε (dm3 mol−1 cm−1): 402(1,810), 318(10,580), 248(15,150).
N–NC), 1256 m ν(C–O), 1937 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 9.9 (s, 1H, CHN), 6.6–7.9 (m, 37H, aromatic), 3.6 (s, 3H, OCH3). UV-Vis (CH3CN, λmax/nm; ε/dm3 mol−1 cm−1): 394(3,140), 318(11,120), 261(15,440).
N–NC), 1292 m ν(C–O), 1946 s ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–7.8 (m, 37H, aromatic), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax/nm; ε/dm3 mol−1 cm−1): 401(3,520), 336(5,590), 264(14,480).
N–NC), 1264 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–7.9 (m, 36H, aromatic), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax /nm; ε/dm3 mol-1 cm-1): 402(3,730), 320(10,920), 253(14,880).
N–NC), 1263 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–7.9 (m, 36H, aromatic), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax (nm); ε(dm3 mol−1 cm−1): 402(3,730), 320(10,920), 253(14,880).
N–NC), 1256 m ν(C–O), 1935 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–8.0 (m, 36H, aromatic), 3.8 (s, 3H, OCH3), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax (nm); ε (dm3 mol−1 cm−1): 398(3,810), 321(10,960), 251(15,320).
O). 1H NMR (400 MHz, DMSO-d6) (δ (ppm)): 9.9 (s, 1H, CHN), 6.8–8.0 (m, 38H, aromatic). UV-Vis (CH3CN, λmax (nm); ε (dm3mol−1 cm−1): 400(3,660), 335(5,720), 264(14,850).
N–NC), 1263 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 9.9 (s, 1H, CHN), 6.9–8.0 (m, 37H, aromatic). UV-Vis (CH3CN, λmax (nm); ε (dm3 mol−1 cm−1): 404(1,780), 320(10,560), 247(15,130).
N–NC), 1263 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 9.9 (s, 1H, CHN), 6.8–8.0 (m, 37H, aromatic). UV-Vis (CH3CN, λmax (nm); ε (dm3 mol−1 cm−1): 402(1,810), 318(10,580), 248(15,150).
N–NC), 1256 m ν(C–O), 1937 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 9.9 (s, 1H, CHN), 6.6–7.9 (m, 37H, aromatic), 3.6 (s, 3H, OCH3). UV-Vis (CH3CN, λmax/nm; ε/dm3 mol−1 cm−1): 394(3,140), 318(11,120), 261(15,440).
N–NC), 1292 m ν(C–O), 1946 s ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–7.8 (m, 37H, aromatic), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax/nm; ε/dm3 mol−1 cm−1): 401(3,520), 336(5,590), 264(14,480).
N–NC), 1264 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–7.9 (m, 36H, aromatic), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax /nm; ε/dm3 mol-1 cm-1): 402(3,730), 320(10,920), 253(14,880).
N–NC), 1263 m ν(C–O), 1946 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–7.9 (m, 36H, aromatic), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax (nm); ε(dm3 mol−1 cm−1): 402(3,730), 320(10,920), 253(14,880).
N–NC), 1256 m ν(C–O), 1935 m ν(CO). 1H NMR (400 MHz, CDCl3) (δ (ppm)): 10.0 (s, 1H, CHN), 6.9–8.0 (m, 36H, aromatic), 3.8 (s, 3H, OCH3), 2.5 (s, 3H, CH3). UV-Vis (CH3CN, λmax (nm); ε (dm3 mol−1 cm−1): 398(3,810), 321(10,960), 251(15,320).
X-ray crystallography
Single crystals of [Ru(L1)(CO)Cl(AsPh3)2] (1) and [Ru(L5)(CO)Cl(AsPh3)2] (5) were grown by slow evaporation of a methanol solution at room temperature. The data collection was carried out using a Bruker AXS Kappa APEX II single crystal X-ray diffractometer using monochromated Mo–Kα radiation (kI = 0.71073 Å). Data was collected at 293 K. The absorption corrections were performed by the multi-scan method using SADABS software.26 Corrections were made for Lorentz and polarization effects. The structures were solved by direct methods (SHELXS 97) and refined by full-matrix least squares on F2 using SHELXL 97.27 All non-hydrogen atoms were refined anisotropically and the hydrogen atoms in these structures were located from the difference Fourier map and constrained to the ideal positions in the refinement procedure. The unit cell parameters were determined by the method of difference vectors using reflections scanned from three different zones of the reciprocal lattice. The intensity data were measured using ω and ϕ scan with a frame width of 0.5°. Frame integration and data reduction were performed using the Bruker SAINT-Plus (Version 7.06a) software.28Typical procedure for the one-pot conversion of aldehydes to amides
To an oven-dried round-bottom flask equipped with magnetic stirring bar was added complex 1 (2.98 × 10−4 mol), aldehyde (5.96 × 10−4 mol), NH2OH·HCl (5.96 × 10−4 mol) and NaHCO3 (5.96 × 10−4 mol), and the mixture was placed under an atmosphere of N2. Dry and degassed MeCN (2 mL) was added and the reaction mixture was refluxed for the time specified under an N2 atmosphere. The reaction was cooled to room temperature and the solvent evaporated. The residue was dissolved in CH2Cl2, filtered and the solvent removed. The crude product was then purified using silica gel chromatography (CHCl3/MeOH) giving the amides in high isolated yields. The products were confirmed by 1H and 13C NMR.Acknowledgements
We sincerely thank the Council of Scientific and Industrial Research (CSIR), New Delhi for financial support [09/475(0140)2008-EMR-I] and for the Senior Research Fellowship for R.N.P. We express sincere thanks to D.S.T-India (F.I.S.T. programme) for the use of the Bruker Smart APEX II diffractometer at the School of Chemistry, Bharathidasan University.References
-
(a) V. Balzani, A. Juris and M. Venturi, Chem. Rev., 1996, 96, 759 CAS
; (b) K. Kalyanasundaram and L. Gratzel, Coord. Chem. Rev., 1998, 177, 347 CAS
-
(a) M. Pagliaro, S. Campestrini and R. Ciriminna, Chem. Soc. Rev., 2005, 34, 837 CAS
-
(a) O. C. P. Beers, M. M. Bouman, C. J. Elsevier, W. J. J. Smeets and A. L. Spek, Inorg. Chem., 1993, 32, 3015 CAS
-
(a) M. Carcelli, C. Pelizzi, G. Pelizzi, P. Mazza and F. Zani, J. Organomet. Chem., 1995, 488, 55 CAS
-
(a) J. C. Galiz, J. C. Rub and J. Edger, Nature, 1955, 34, 176 Search PubMed
-
(a) R. Dinda, P. Sengupta, S. Ghosh, H. Mayer-Figge and W. S. Sheldrick, J. Chem. Soc., Dalton Trans., 2002, 4434 CAS
-
(a) S. N. Pal and S. Pal, J. Chem. Soc., Dalton Trans., 2002, 2102 CAS
- D. Mishra, S. Naskar, A. J. Blake and S. K. Chattopadhyay, Inorg. Chim. Acta, 2007, 360, 2291 CAS
-
(a) V. Mahalingam, N. Chitrapriya, F. R. Fronczek and K. Natarajan, Polyhedron, 2008, 27, 1917 CAS
-
(a)
M. G. Loudon, in Organic chemistry, Oxford University Press, New York, NY, 2002, p. 982 Search PubMed
-
(a) C. A. G. N. Montalbetti and C. Falque, Tetrahedron, 2005, 61, 10827 CAS
- V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1 CAS
- H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., 2007, 119, 5294 Search PubMed
- C. L. Allen, C. Burel and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 2724 CAS
- D. Gnanamgari and R. H. Crabtree, Organometallics, 2009, 28, 922 CAS
- J. F. Hull, S. T. Hilton and R. H. Crabtree, Inorg. Chim. Acta, 2010, 363, 1243 CAS
- R. R. Gowda and D. Chakraborty, Eur. J. Org. Chem., 2011, 2226 CAS
-
(a) R. N. Prabhu, D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2009, 694, 4170 CAS
- R. Raveendran and S. Pal, J. Organomet. Chem., 2007, 692, 824 CAS
-
(a) M. Cao, L. V. Do, N. W. Hoffman, M.-L. Kwan, J. K. Little, J. M. McGilvray, C. B. Morris, B. C. Söderberg and A. Wierzbicki, Organometallics, 2001, 20, 2270 CAS
-
(a) K. N. Kumar and R. Ramesh, Spectrochim. Acta, Part A, 2004, 60, 2913 Search PubMed
- S. Kannan, M. Sivagamasundari, R. Ramesh and Y. Liu, J. Organomet. Chem., 2008, 693, 2251 CAS
-
(a) K. N. Kumar, R. Ramesh and Y. Liu, J. Inorg. Biochem., 2006, 100, 18 CAS
-
A. I. Vogel, Test book of practical organic chemistry, Longman, London, 5th edn, 1989, p. 395 Search PubMed
- R. A. Sanchez-delgado, W. Y. Lee, S. R. Choi, Y. Cho and M. J. Jun, Transition Met. Chem., 1991, 16, 241 CAS
- F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25 CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 Search PubMed
- Bruker-Nonius ( 2004), APEX-II and SAINT-Plus (Version 7.06a), Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra for all the complexes, 1H and 13C NMR spectra for all the amides. CCDC reference number 816486 (for 1) and 829046 (for 5). See DOI: 10.1039/c2ra20382k |
| This journal is © The Royal Society of Chemistry 2012 |