Bis-Sonogashira cross-coupling: an expeditious approach towards long-chain, phenylene-modified 1,ω-diols†
Simon
Drescher
*ab,
Susan
Becker
a,
Bodo
Dobner
a and
Alfred
Blume
b
aInstitute of Pharmacy, Wolfgang-Langenbeck-Str. 4, 06120, Halle/Saale, Germany. E-mail: simon.drescher@pharmazie.uni-halle.de; Fax: +49(0) 345 5527026; Tel: +49(0) 345 5525196
bInstitute of Chemistry, von-Danckelmann-Platz 4, 06120, Halle/Saale, Germany
First published on 9th March 2012
Abstract
The synthesis of long-chain, phenylene-modified 1,ω-diols can be effectively performed in 60% overall yield using the bis-Sonogashira cross-coupling with 6 mol% of PdCl2(PPh3)2 and tetrabutylammonium fluoride in a copper-, amine- and solvent-free setting followed by hydrogenation.
An expeditious and highly efficient method for the synthesis of long-chain 1,ω-diols is of high importance in supramolecular chemistry. Especially in the field of bipolar lipid (bolaamphiphile1) synthesis, these diols play an important role since they represent the hydrophobic part of this class of lipids. These bolaamphiphiles were originally found in the membrane lipids of certain species of Archaea.2 In particular, the bipolar archaebacterial model lipids3 are of great interest since their thermal and chemical stability as well as their capability to form closed vesicles are outstanding properties. These properties make this class of amphiphiles applicable in biotechnology, materials science and pharmacy.4 For instance, thermally and chemically stable liposomes made from archaebacterial bolaamphiphiles (archaeosomes) are an alternative to conventional liposomes in applications as a drug delivery system.5
Long-chain, unmodified 1,ω-diols with 22 or more carbon atoms have already been synthesised using established multistep procedures such as (i) bis-acylation of cyclic enamines with dicarboxylic acid dichlorides, (ii) hydrolysis of enamino ketones and subsequent ring opening, (iii) Wolff–Kishner reduction of bis(oxoacid)s and subsequent reduction with lithium aluminium hydride, or (iv) double Wittig reaction with bis(phosphorylide)s and ω-functionalised aldehydes.6 In our group, we use the Grignard reaction under catalysis of dilithium tetrachlorocuprate(II)7 for the synthesis of long-chain 1,ω-diols. This coupling reaction can be designed as a homo-coupling8 or a bis-coupling reaction.9 For the synthesis of the entitled phenylene-modified compounds we previously used the Grignard reaction in a stepwise homo-coupling procedure since the simultaneous bis-coupling reaction resulted in the formation of by-products, which were impossible to separate.10 Although this homo-coupling approach resulted in pure products and provided the opportunity for the synthesis of unsymmetrical bolalipids, it is a very time-consuming synthetic pathway.10 Therefore, the expeditious Sonogashira cross-coupling reaction is a rational alternative for the preparation of such phenylene-modified diols.
Sonogashira cross-coupling11 is one of the most powerful and straightforward methods for the formation of carbon–carbon bonds in organic synthesis.12 Usually, the Pd-catalysed reaction conditions are mild, and many reactions can be carried out at ambient temperatures. However, the Sonogashira reaction often generates homo-coupling products of terminal alkynes,13 difficult to separate from the target products due to similar chromatographic properties.14 Since the formation of these by-products is due to the addition of the co-catalyst CuI, numerous copper-free catalyst systems were developed.15 Besides the ‘homo’-Sonogashira, bis-Sonogashira cross-coupling reactions have been developed using either middle-chain alkynes (up to 11 carbon atoms) and vinyl halides16 or aromatic (benzene or pyridine) dialkynes and aryl iodides.17 However, all these approaches used copper(I), bearing the risk of the formation of homo-coupling side-products. This problem has to be considered in the synthesis presented here, because long-chain 1,ω-diols cannot be separated from their phenylene-modified analogues.10
The present work describes the preparation of phenylene-modified 1,ω-diols starting from benzene dihalides 1 and the long-chain unprotected alkynol 2, including different substitution patterns (Scheme 1).
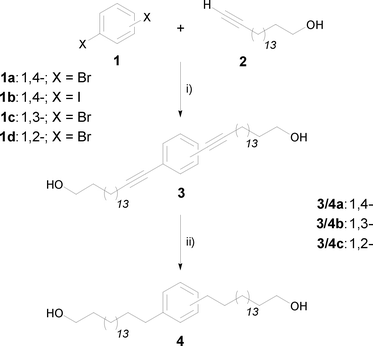 | ||
| Scheme 1 Reagents and conditions: (i) PdCl2(PPh3)2, TBAF·3H2O, 1 h, 80 °C (43–63%); (ii) H2, Pd(OH)2/C (20%), heptane/ethyl acetate/ethanol, 10 atm, 18 h, rt (90–95%). | ||
Due to the very low solubility of compound 2 in common solvents used for Sonogashira coupling reactions, we investigated DMSO and water combined with a micellar system as solvents, and also solvent-free systems. Furthermore, several Pd-catalysts were tested (see Table 1).
| Entry | Dihalide (mmol) | 2 (mmol) | Pd-catalyst (mol%) | Solvent | Additives (mmol) | Temp. (°C) | Time (h) | Yield (%)b |
|---|---|---|---|---|---|---|---|---|
a Conditions: all reactions were carried out under an argon atmosphere.
b Isolated yields after chromatography (MPLC).
c TBAB: tetra-n-butylammonium bromide.
d Product was contaminated with homo-coupling by-product.
e X-Phos: 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
f PTS: polyoxyethanyl-α-tocopheryl sebacate (3 wt% in H2O; Sigma Aldrich catalogue no. 698![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 717).
g TBAF: tetra-n-butylammonium fluoride trihydrate. 717).
g TBAF: tetra-n-butylammonium fluoride trihydrate.
|
||||||||
| 1 | 1a (0.5) | 1.2 | Pd(OAc)2 (3) | DMSO | K2CO3 (2), TBABc (1) | 55 | 6 | 10d |
| 2 | 1b (0.5) | 1.2 | Pd(OAc)2 (3) | DMSO | K2CO3 (2), TBABc (1) | 65 | 4.5 | 9 |
| 3 | 1a (0.5) | 1.2 | Pd(CH2CN)2Cl2 (4.8), X-Phose (10.4) | H2O | PTSf (3 mL), Et3N (2.2) | rt | 96 | 14 |
| 4 | 1a (0.5) | 1.2 | Pd(CH2CN)2Cl2 (2.4), X-Phose (5.2) | H2O | PTSf (3 mL), Et3N (2.2) | 45 | 8 | 10 |
| 5 | 1a (0.5) | 1.2 | PdCl2(PPh3)2 (3) | H2O | PTSf (3 mL), Et3N (2.2) | rt | 24 | 10 |
| 6 | 1a (1.0) | 2.4 | PdCl2(PPh3)2 (6) | None | TBAFg (3) | 60 | 1 | 42 |
| 7 | 1a (0.8) | 1.9 | PdCl2(PPh3)2 (6) | None | TBAFg (2.4) | 80 | 1 | 63 |
| 8 | 1a (0.5) | 1.2 | PdCl2(PPh3)2 (6) | None | TBAFg (1.5) | 80 | 2 | 56 |
| 9 | 1b (0.8) | 1.9 | PdCl2(PPh3)2 (6) | None | TBAFg (2.4) | 80 | 1 | 45 |
| 10 | 1c (0.8) | 1.9 | PdCl2(PPh3)2 (6) | None | TBAFg (2.4) | 80 | 1 | 61 |
| 11 | 1d (0.8) | 1.9 | PdCl2(PPh3)2 (6) | None | TBAFg (2.4) | 80 | 1 | 43 |
At first, we adapted a method described by Li et al.18 using Pd(OAc)2 as catalyst combined with the relatively strong base potassium carbonate and tetra-n-butylammonium bromide (TBAB) at elevated temperatures (entries 1, 2). In addition, we changed the solvent from ethanol to DMSO in order to obtain a better solubility of compound 2. However, the desired product (3a) could be obtained only in marginal yields of about 10%. Unexpectedly, while using the bromo derivative 1a, we detected the homo-coupling product of the terminal alkynol 2, which could not be separated from the product (entry 1). When we used the iodide 1b instead (entry 2), the formation of the by-product could be avoided.
In a next step (entries 3, 4), we applied the method described by Lipshutz and co-workers15a using the micelle-forming amphiphile PTS19 in aqueous medium, together with Et3N as a soluble amine and X-Phos20 as ligand in the presence of catalytic amounts of PdCl2(CH3CN)2. The rationale behind this approach was that the use of a micellar system21 might enhance the solubility of the educts and, hence, should trigger the Sonogashira cross-coupling reaction. However, both reactions (entries 3, 4), using different amounts of catalyst and ligand, respectively, and working at different temperatures, gave only low yields of the desired product. In contrast with entry 1, the formation of the homo-coupling by-product was not detected. The use of PdCl2(PPh3)2 instead of PdCl2(CH3CN)2 + X-Phos (entry 5) led to no improvement regarding yield. In addition, the PTS/water system combined with the long-chain alkynol caused problems during the purification process since a gel-like suspension was formed.
The next reaction conditions tested (entries 6–11) were inspired by the work of Mori and co-workers:22 they used TBAF as an activator for the amine-free Pd-catalysed Sonogashira cross-coupling reaction. However, organic solvents such as THF were still required. Based on this procedure, Liang et al.23 used 3 mol% of PdCl2(PPh3)2 combined with TBAF under solvent-free conditions: For the long-chain alkyne (decyne) and substituted aryl mono-bromides and mono-chlorides, respectively, they obtained 51–98% isolated yields within 1 to 25 h of reaction.23
In our experiments, the reaction of dibromo benzene 1a with the alkynol 2 in a 1.2-fold excess using 6 mol% of PdCl2(PPh3)2 and 3 equivalents of TBAF (entry 6), and without any solvent as medium, afforded 42% of the desired product 3a.24 An increase in reaction temperature from 60 to 80 °C (entry 7) resulted in an increased yield (63%), whereas an increase in reaction time (entry 8) caused a slight decrease in yield. Unexpectedly, the use of diiodo benzene 1b (entry 9) resulted in lower yields of the desired product combined with a higher amount of unknown by-products. This might be due to the higher reactivity of the iodine compound compared to the bromine counterpart. A change in the substitution pattern of the bromo derivative 1a caused only small variations in product composition: the lower yield while using the ortho derivative 1d (entry 11) is due to steric hindrance during the bis-Sonogashira cross-coupling reaction.
In all reactions using the PdCl2(PPh3)2/TBAF system no homo-coupling products of the terminal alkynol were observed. The mono-coupling Sonogashira product, which always occurred in the cross-coupling reaction in 5–20%, could successfully be separated by middle pressure liquid chromatography (MPLC).
Finally, the hydrogenation of the triple bonds of compounds 3 using a solvent mixture of heptane, ethyl acetate and ethanol, to ensure complete dissolution of the product, and Pd(OH)2 on carbon resulted in the formation of the target products 4. For a complete conversion it is necessary to use increased hydrogen pressure (up to 10 atm) over several hours. Otherwise, mixtures of products with double bonds are formed.
In summary, we have developed a general synthetic approach for the expeditious preparation of long-chain, phenylene-modified 1,ω-diols. We found that the bis-Sonogashira cross-coupling reaction of aryl dibromides with terminal alkynols, using PdCl2(PPh3)2 and TBAF, followed by hydrogenation is the method of choice, which circumvents the formation of homo-coupling products. This synthetic approach is also applicable for other chain lengths and/or biphenyl/terphenyl core-structures, giving rise to a huge variety of different, phenylene-modified 1,ω-diols important in supramolecular chemistry. The synthesis as well as the physicochemical characterisation of the corresponding bolaamphiphiles are currently underway.
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (projects Bl 182/19-3 and Do 463/4-2).References
- J.-H. Fuhrhop and T. Wang, Chem. Rev., 2004, 104, 2901 CrossRef CAS.
- (a) T. A. Langworthy, Biochim. Biophys. Acta, Lipids Lipid Metab., 1977, 487, 37 CrossRef CAS; (b) M. De Rosa, E. Esposito, A. Gambacorta, B. Nicolaus and J. D. Bu'Lock, Phytochemistry, 1980, 19, 827 CrossRef CAS.
- (a) T. Benvegnu, M. Brard and D. Plusquellec, Curr. Opin. Colloid Interface Sci., 2004, 8, 469 CrossRef CAS; (b) A. Meister and A. Blume, Curr. Opin. Colloid Interface Sci., 2007, 12, 138 CrossRef CAS.
- (a) B. A. Cornell, V. B. L. Braach-Maksvytis, L. G. King, P. D. J. Osmann, B. Raguse, L. Wieczorek and R. J. Pace, Nature, 1997, 387, 580 CrossRef CAS; (b) U. Bakowsky, U. Rothe, E. Antonopoulos, T. Martini, L. Henkel and H. J. Freisleben, Chem. Phys. Lipids, 2000, 105, 31 CrossRef CAS; (c) T. Benvegnu, G. Réthoré, M. Brard, W. Richter and D. Plusquellec, Chem. Commun., 2005, 5536 RSC; (d) G. Réthoré, T. Montier, T. Le Gall, P. Delépine, S. Cammas-Marion, L. Lemiègre, P. Lehn and T. Benvegnu, Chem. Commun., 2007, 2054 Search PubMed; (e) G. P. Patel, A. Ponce, H. Zhou and W. Chen, Int. J. Toxicol., 2008, 27, 329 CrossRef CAS; (f) D. A. Brown, B. Venegas, P. H. Cooke, V. English and P. L.-G. Chong, Chem. Phys. Lipids, 2009, 159, 95 CrossRef CAS.
- G. D. Sprott, D. L. Tolson and G. B. Patel, FEMS Microbiol. Lett., 1997, 154, 17 CrossRef CAS.
- (a) S. Hünig, E. Lücke and W. Brenninger, in Org. Synth. Coll., vol. 5, New York, 1973, p. 533 Search PubMed; (b) I. Grechishnikova, L. B.-A. Johansson and J. G. Molotkovsky, Chem. Phys. Lipids, 1996, 81, 87 CrossRef CAS; (c) R. McKiernan, S. P. Gido and J. Penelle, Polymer, 2002, 43, 3007 CrossRef CAS.
- M. Tamura and J. Kochi, Synthesis, 1971, 303 CrossRef CAS.
- (a) U. F. Heiser and B. Dobner, Chem. Commun., 1996, 2025 RSC; (b) U. F. Heiser and B. Dobner, J. Chem. Soc., Perkin Trans. 1, 1997, 809 RSC.
- (a) K. Köhler, G. Förster, A. Hauser, B. Dobner, U. F. Heiser, F. Ziethe, W. Richter, F. Steiniger, M. Drechsler, H. Stettin and A. Blume, J. Am. Chem. Soc., 2004, 126, 16804 CrossRef; (b) K. Köhler, G. Förster, A. Hauser, B. Dobner, U. F. Heiser, F. Ziethe, W. Richter, F. Steiniger, M. Drechsler, H. Stettin and A. Blume, Angew. Chem., Int. Ed., 2004, 43, 245 CrossRef; (c) S. Drescher, A. Meister, A. Blume, G. Karlsson, M. Almgren and B. Dobner, Chem.–Eur. J., 2007, 13, 5300 CrossRef CAS; (d) A. Meister, S. Drescher, G. Karlsson, G. Hause, U. Baumeister, G. Hempel, V. M. Garamus, B. Dobner and A. Blume, Soft Matter, 2010, 6, 1317 RSC.
- S. Drescher, S. Sonnenberger and B. Dobner, unpublished results.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- (a) H. Doucet and J.-C. Hierso, Angew. Chem., 2007, 119, 850 CrossRef; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS; (c) E. Negishi and L. Anaytasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS; (d) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS; (e) M. Erdelyi and A. Gogoll, J. Org. Chem., 2001, 66, 4165 CrossRef CAS; (f) T. Hundertmark, A. F. Littke, S. L. Buchwald and G. C. Fu, Org. Lett., 2000, 2, 1729 CrossRef CAS.
- (a) P. Siemsen, R. C. Livingston and F. Dieterich, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS; (b) J.-H. Li, Y. Liang and X.-D. Zhang, Tetrahedron, 2005, 61, 1903 CrossRef CAS.
- H. Kukula, S. Veit and A. Godt, Eur. J. Org. Chem., 1999, 277 CrossRef CAS.
- (a) B. H. Lipshutz, D. W. Chung and B. Rich, Org. Lett., 2008, 10, 3793 CrossRef CAS; (b) A. Soheili, J. Albaneze-Walker, J. A. Murry, P. G. Dormer and D. L. Hughes, Org. Lett., 2003, 5, 4191 CrossRef CAS; (c) Y. Ma, C. Song, W. Jiang, Q. Wu, Y. Wang, X. Liu and M. B. Andrus, Org. Lett., 2003, 5, 3317 CrossRef CAS; (d) K. Heuzé, D. Méry, D. Gauss and D. Astruc, Chem. Commun., 2003, 2274 RSC; (e) V. P. W. Böhm and W. A. Herrmann, Eur. J. Org. Chem., 2000, 3679 CrossRef.
- (a) E. Quesada, A. U. Acuna and F. Amat-Guerri, Eur. J. Org. Chem., 2003, 1308 CrossRef CAS; (b) E. Quesada, A. U. Acuna and F. Ama-Guerri, Angew. Chem., Int. Ed., 2001, 40, 2095 CrossRef CAS.
- (a) T. Shoji, S. Ito, K. Toyota, M. Yasunami and N. Morita, Chem.–Eur. J., 2008, 14, 8398 CrossRef CAS; (b) K. Kobayashi and N. Kobayashi, J. Org. Chem., 2004, 69, 2487 CrossRef CAS; (c) M. Akhtaruzzaman, M. Tomura and B. M. Zaman, J. Org. Chem., 2002, 67, 7813 CrossRef CAS; (d) A. Khatyr and R. Ziessel, J. Org. Chem., 2000, 65, 7814 CrossRef CAS; (e) P. N. W. Baxter, J. Org. Chem., 2000, 65, 1257 CrossRef CAS.
- P. Li, L. Wang and H. Li, Tetrahedron, 2005, 61, 8633 CrossRef CAS.
- H. Borowy-Borowski, M. Sikorska-Walker and P. R. Walter, U.S. Patent 6,045,826, April 4, 2000 Search PubMed.
- D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993 CrossRef CAS.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174 CrossRef CAS.
- (a) A. Mori, T. Shimada, T. Kondo and A. Sekiguchi, Synlett, 2001, 649 CAS; (b) A. Mori, J. Kawashima, T. Shimada, M. Suguro, K. HIrabayashi and Y. Nishihara, Org. Lett., 2000, 2, 2935 CrossRef CAS.
- Y. Liang, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2006, 71, 379 CrossRef CAS.
- Experimental procedure for a typical bis-Sonogashira cross-coupling reaction: an oven-dried flask was filled with aryl dihalide 1 (0.5–1.0 mmol), alkynol 2 (1.2–2.4 mmol), PdCl2(PPh3)2 (6 mol%) and TBAF·3H2O (3 equiv.) under an argon atmosphere. The mixture was subsequently stirred at 80 °C for 1 h until consumption of the starting material was complete. Afterwards, 30 mL H2O was added and the mixture was extracted with chloroform (3 × 20 mL). The combined organic layers were washed with brine (20 mL), and water (20 mL), dried over sodium sulfate, and evaporated. The residue was purified by middle pressure liquid chromatography (MPLC) using chloroform/diethyl ether as eluents and a gradient technique to afford the diols 3 as white to light yellow crystalline powders. For the subsequent hydrogenation of the triple bonds, diols 3 were dissolved in heptane/ethyl acetate/ethanol (100 mL, 3/1/1, v/v/v) and Pd(OH)2 (20% on carbon, 50–100 mg) was added. The mixture was stirred under hydrogen (10 atm) at room temperature for 18 h. Afterwards, the catalyst was removed by filtration and the solvent was evaporated in vacuo to give the diols 4 as white crystalline powders.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. NMR spectra of isolated compounds. See DOI: 10.1039/c2ra20411h |
| This journal is © The Royal Society of Chemistry 2012 |