DOI:
10.1039/C2RA20451G
(Review Article)
RSC Adv., 2012,
2, 8557-8578
Smart polymer brushes and their emerging applications
Received
12th March 2012
, Accepted 3rd June 2012
First published on 13th June 2012
Abstract
Polymer brushes with stimuli-responsive properties, also known as smart polymer brushes (SPBs), have attracted substantial research interest in the past few years because they offer a wealth of opportunities to design responsive materials triggered by external stimuli, and have a number of applications in many areas in science and technology. Changing the length, chemical composition, architecture, and topology of the chains allows a change in response mechanisms and rates. This review introduces some key strategies for preparing polymer brushes and discusses various smart polymer brush surfaces and the recent developments on route to application. Current and future challenges in this field of smart polymer brushes are addressed.
1 Introduction
Polymer brushes can be defined as long-chain macromolecules that are attached with the anchor sites to a surface1 or these graft copolymers in which multiple polymer chains are grafted to a linear polymer. They are ensembles of sufficiently densely packed polymer chains.2–4 Smart macromolecular brushes are also called as stimuli-responsive polymer brushes in some literatures, whose physicochemical properties and shape usually show rapid and reversible changes in response to small changes of their environment (e.g., pH, temperature, salt concentration, electric potential, solvent and surrounding media).5–7 Smart polymer brushes (SPBs) in the past few years have attracted substantial research interest because these responses can be changed or tuned in an accurate and predictable manner by using an external stimulus, and these smart polymer brush surfaces play an important role in a diverse range of applications to many areas in science and technology.5,8–16 Smart polymeric-based devices and surfaces that reversibly alter their physico-chemical characteristics in response to their different environment are the center of many studies related to the development of materials and concepts in a broad-range of multi-disciplinary fields, which include responsive biointerfaces that are functionally similar to natural surfaces,17,18 controlled drug-delivery and release systems,12,19 anti-fog surfaces and sensor devices,20,21 coatings that are capable of interacting and responding to their environment,22,23 composite materials that actuate and mimic the action of muscles,24 and thin films and particles that are capable of sensing very small concentrations of analytes.25,26
This article concentrates on SPBs that are capable of conformational and chemical changes caused by the external stimuli signal from their environment. These changes are accompanied by variations in the physical properties of polymer. Here, we introduce the key strategies for preparing SPBs, discuss different smart polymer brush surfaces and review the recent developments on the route to application. Finally, challenges in making synthetic systems capable of responding to stimuli in a controllable and predictable manner and future prospectives are examined.
2 The classification and fabricating strategy of polymer brush
Polymer brushes can be defined as long-chain polymer molecules that are attached with one or with a few anchor sites to a surface or a linear polymer.1,27–30 In polymer brushes, the distance between grafting points is smaller than the chain end-to-end distance. The main chain is commonly referred to as the backbone and the branches as the side chains. If the length of the backbone is significantly longer than that of the side chains, intramolecular excluded volume effects cause the polymer to adopt a cylindrical shape with the backbone polymer in the core, from which the side chains emanate radially.31–40 Conversely, macromolecular brushes with backbones on the order of the length of the side chains generally adopt compact, spherical dimensions that resemble star polymers.41 Many synthetic surface-tethered smart polymers, known as SPBs, respond to small external changes in their environment with dramatic property variations.12,42–46 These polymers can have different composition and architecture, including not only homo-polymers, but also statistical/block copolymers,47 grafting copolymers and molecular brushes. Due to densely grafted side chains on a linear polymer main chain, they can be imaged as single molecules by atomic force microscopy (AFM) (Fig. 1a) similar to photographs that are found in a variety of places within the body (Fig. 1b). Molecular brushes have been proposed as synthetic substitutes in order to understand the architecture-property relationship.
 |
| | Fig. 1 AFM images of molecular brushes: (a) molecular brushes with poly (n-butyl acrylate) (PBA) side chains, and (b) proteoglycans having a protein backbone with glucosaminoglycan side chains.27,221 | |
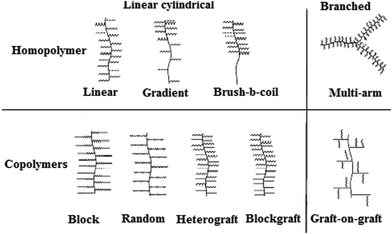 |
| | Fig. 2 Classification of molecular brushes by branching topologies and chemical composition of side chains.65,66 | |
2.1 The types of polymer brush
Based on the chemical compositions in the side chains, molecular brushes can be divided into two categories: (1) brushes with homopolymer side chains: linear brushes, gradient brushes with a gradient in grafting density along the copolymer backbone,48–50 and brush–block–coil copolymers 51,52 and (2) brushes with copolymer side chains: brushes with random/block copolymer side chains,53–60 heterografted brushes,61,62 and AB-type brush (block brush) copolymers28,63,64etc. While the simplest molecular brushes with one type of homopolymer side chain have been extensively studied, copolymer side chain brushes have also been prepared. Besides the linear cylindrical shape of molecular brushes, special types of brushes with various branching topologies have been synthesized such as star-like multiarm structures.65–67
2.2 The fabricating strategy of polymer brush
Fixing or anchoring one end of the polymer chain onto a surface, i.e. tethering of the polymer, is one popular method to obtain polymer brushes. Tethering of the polymer to the surface, known as polymer brushes, can be performed either by physical adsorption or covalent attachment. Covalent attachment is often preferred due to the inherent resistance to degradation by temperature or solvents. Atom transfer radical polymerization (ATRP),68–73 stable free radical polymerization (SFRP),74,75 and reversible addition fragmentation chain transfer (RAFT) polymerization techniques76–80 have been the most successful methods. The tethering should be sufficiently dense so that the polymer chains can adopt a defined, stretched chain conformation, which significantly differs from the random-walk conformation of free polymer chains in solution or in conventional solution cast polymer coatings. Controlled/living radical polymerization (CRP) has also been employed for the preparation of molecular brushes,81 which has provided a versatile route for the synthesis of well-controlled polymers with predetermined molecular weight (MW), narrow molecular weight distribution (MWD), various architectures, and useful end-functionalities.82–87 We can sum up the strategy to attach polymers to a substrate using the three approaches described below.
2.2.1 Surface grafting.
Numerous SPBs were developed by chemical grafting. Generally, as shown schematically in Fig. 3, there are three methods for the synthesis of molecular brushes: “grafting to”, “grafting through” (polymerization of macromonomers), and “grafting from” (grafting the side chains from the backbone).29,88–133
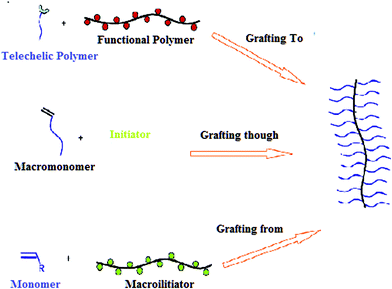 |
| | Fig. 3 Grafting methods for preparing molecular brushes by “grafting to”, “grafting through”, and “grafting from”. “Grafting to” involves the coupling of individual side chains to a common backbone polymer. “Grafting through” consists of the polymerization of macromonomers. “Grafting from” involves growth of the side chains from a well-defined backbone macroinitiator.299 | |
In the “grafting-to” approach, both backbone and side chains are prepared separately, the reaction of end-functional polymers with a polymer backbone precursor containing complimentary functionality on each monomer unit forms covalently bonded layers. Therefore, functional macromolecular chain can tether to the surface or substrate with reactive groups by coupling reaction, radical addition, condensation reaction, etc. In addition, the end-functional polymer chain may attach to the active surface through physicochemical adsorption. However, slow diffusion of the bulky side chains hinders preparation of brushes with high grafting density. In the “grafting from” method, a macroinitiator with a predetermined number of initiation sites is prepared, followed by grafting the side chains from the macroinitiator, therefore this kind of polymerization belongs to surface-initiated polymerization (SIP). Growth of side chains has been mainly achieved via ATRP initiated by pendant bromoester groups on a polymer backbone or on a substrate. In ATRP and other CRP, initiator usually plays a key role for the growth of polymer brush. The popular initiator mainly includes transition metal solid/pyridine ligand; α-organic bromide (such as α-bromoketone, etc.). The “grafting from” or surface-initiated polymerization method usually produces polymer brushes with a higher grafting density and a greater thickness, but it requires the immobilization of an initiator on the surface first. These mechanisms are suitable for molecular brush synthesis via grafting from. The grafting through route involves the polymerization of macromonomers–polymers with polymerizable end groups—“through” their terminal functionality. This method can obtain polymer brushes with high grafting density, assuming a sufficient purity of the original macromonomer. It is difficult, however, to synthesize molecular brushes with a high degree of polymerization (DP) and low polydispersity because of the inherently low concentration of polymerizable groups and the steric hindrance of side chains.
2.2.2 Self-assembled monolayers (SAMs).
In general, the attachment of surface-bound initiators onto the substrate needs to be performed prior to SIP studies. Particularly common is the attachment of surface-bound initiators via formation of SAMs,134,135 which form spontaneously by the adsorption of an active material onto a surface, and possess important properties of self-organization and adaptability to a number of technologically relevant surface substrates. Molecular structures forming SAMs can be classified into three features: (1) a headgroup which strongly binds to the substrate; (2) an endgroup that allows the introduction of a variety of organic functionalities to be incorporated into a monomolecular film; (3) and a spacer unit that connects the headgroup and endgroup and strongly affects the intermolecular separation, molecular orientation and the degree of order in surface. As such the properties of a SAM (thickness, structure, surface energy, stability) can be easily controlled and specific functionalities can be introduced into the building blocks, monomers can be polymerized from surface-anchored initiators generally immobilized by the SAM technique.17,136–138 Therefore, SAMs offer ease of preparation and versatile surface chemistry, polymer brushes can be produced by SIP techniques with a better control over surface coverage, thickness and composition.
2.2.3 The combined layer-by-layer (LbL) and SIP techniques.
Surface tethered polymer chains by chemisorption method can overcome the weaker adhesion to the surface inherent with the physisorption method. In order to ensure that the surface tethered chains grow in the brush regime, a sufficiently high initiator density is required.139 In a previous publication, Advincula's group demonstrated the fabrication of intelligent surfaces based on pH and temperature responsive polymer brushes utilizing the combined LbL and SI-ATRP techniques.140 Relatively thick and dense poly (n-isopropylacrylamide) (PNIPAM) brushes were produced by having multiple layers of the ATRP macroinitiators deposited. The sole deposition of polyelectrolyte macroinitiators and the subsequent polymerization of dense poly (2-hydroxyethyl methacrylate) (PHEMA) polymer brushes have also been reported.141 They have utilized the LbL technique on flat substrates to electrostatically adsorb oppositely charged macroinitiators onto a functionalized substrate, achieving high initiator densities compared to SAMs. Novel films exhibiting properties of low surface energy were prepared with these LbL brush films (Fig. 4).
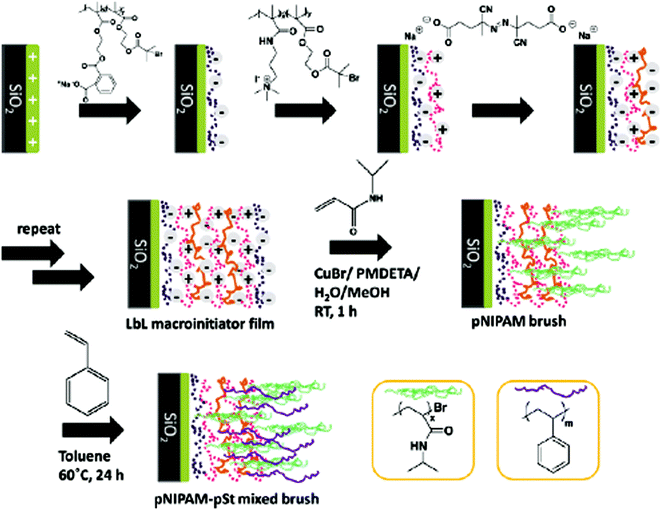 |
| | Fig. 4 Brush surface fabrication via the layer-by-layer (LbL) technique.297 | |
3 Smart polymer brush surfaces
With the application of an external stimulus, smart polymer brush surfaces may be capable of physical and chemical changes. These changes are accompanied by variations in energy of the brush system owing to reconstruction of the polymer chains tethered to the surface.142
3.1 The energetic requirements of smart polymer surface
Due to the anchoring of one end of the polymeric segment to the surface, restricted freedom of movement is “transmitted” along the chain. The energy required for the segments further away from the anchoring point to respond is a function of the distance from the surface.143 Those segments close to the anchoring points of surface, the transitions from A to B needs relatively higher energy input (ΔESR(surface)) because more space and free volume are available further away from the anchoring points. Fig. 5 illustrates the relationship between the surface layer equilibrium energy (z axis), stimulus energy input (x axis), and a physical/chemical response to a stimulus (y axis). The free energy in a polymer brush system is used to evaluate polymer chain or segment transfers from one state to another state spontaneously. Generally, stimuli-responsiveness at polymeric surfaces is an entropy (ΔS) driven process,144 in which the disorder of anchored chains (ΔS) has greater contributions to the free energy (ΔG) values than the conformational changes resulting from the enthapic component of the free energy (ΔH).
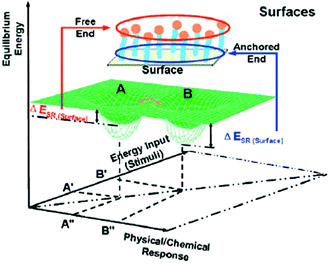 |
| | Fig. 5 The relationship between equilibrium energy, stimuli energy input, and physical/ chemical response.143 | |
3.2 The main stimuli-responsive types of smart polymer brush
The increasing need for “smartness” in biomedical and engineering materials has generated a growing interest for synthetic polymers that exhibit environmentally responsive behavior. One area of substantial research on stimuli-responsive systems contains the simplest homopolymers that undergo reversible conformational changes in response to external stimuli such as solvent, pH, temperature, and so on, leading to changes in physicochemical properties of both materials and single molecules.
3.2.1 Solvent-responsive polymer brushes.
In scaling theory and self-consistent field theory145 the thickness of the polymer brush is linearly proportional to the degree of polymerization N (but not obviously in a bad solvent). Experiments proved that this relationship retains unchanged for solvents of different quality.146 However, the prefactor depends on solvent quality and grafting density of polymer chains. Besides the chain constitution, the grafting density is the parameter which affects conformational responsiveness of polymer brushes.
For most polymers, the macromolecular chain can stretch freely and become coil-like in good and bad solvents, respectively. This will lead to a separation tendency among different polymer segments, i.e. phase segregation of the polymer blend. The solvent quality is capable of affecting polymer chain conformation. Whichever, the mixed polymer brush or block polymer brush, they are capable of switching surface wettability due to a reversible, solvent-induced phase segregation of the brush. This solvent selectivity alters the brush morphology and enables control over which phase becomes exposed to the external environment.147–151 For example, a mixed polymer brush prepared from polystyrene (PS) and poly (2-vinylpyridine) (P2VP)152 changed the surface composition and wetting behavior after treatment in different solvents. The contact-angle change was found to be strongly amplified on a rough surface where the wetting properties switched from complete wetting to ultrahydrophobic behaviour.153 It was shown that a mixed brush made of polystyrene and poly (methylmethacrylate) can induce the local motion (in the range of a few nanometres) of adsorbed nanometre-scale objects through solvent-induced topographical variations of the brush surface.154 A poly (ethyleneimine)–poly (dimethylsiloxane) mixed brush switched spontaneously from the hydrophilic state in water to the hydrophobic one in air.155 This adaptive behaviour of mixed brushes was used to develop materials with poor adhesion in a changeable environment. The surface coatings that were fabricated from mixed poly ethylene oxide (PEO)–poly (dimethylsiloxane) brushes156 were adaptive to liquid and vapour environments so that the surfaces were spontaneously transformed to non-sticky states in air and in water. This behavior was observed on several cycles of exposure of the samples to air and aqueous solutions.
Single molecular conformation transition of brushes by solvent treatments involves the preparation of core–shell molecular brushes in which side chains are composed of hydrophobic–hydrophilic block copolymers. Müller157 synthesized molecular brushes with PS-b-PtBA, PtBA-b-PS and PtBA-b-PBA, PBA-b-PtBA side chains, followed by hydrolysis of the t-butyl groups, which led to acrylic acid units.158,159 These brushes are interesting since they resemble a unimolecular micelle of cylindrical shape. As a result, they are not susceptible to dissociation on dilution while conventional multimolecular micelles dissociate below the critical micelle concentration (CMC). Due to the responsive nature of the PAA blocks toward different kinds of solvent, conformational transitions were observed as a function of solvent quality. Gallyamov et al.160 studied solvent-induced length variation of molecular brushes. They found that the length of brushes varies with the solvent quality of the side chains: the lengths of the backbone and side chains become shorter in a poor solvent as compared to a good solvent.
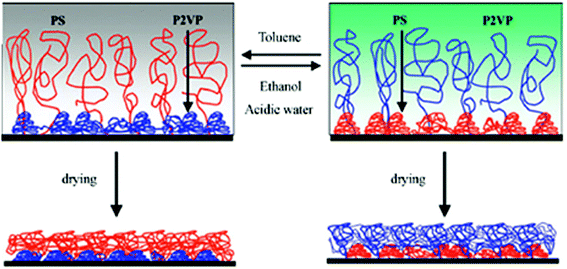
Vyas et al.148 used binary polystyrene (PS)–poly (2-vinylpyridine) (P2VP) brushes to switch between several phase-segregated morphologies and thus affect the chemical composition of the brush top layer (Fig. 6). The switching process can be controlled by treating the binary brushes with selective solvents, such as toluene for PS or acidic water for P2VP. When these brushes are treated with toluene, PS chains swell and stretch away from the surface to preferentially occupy the outer-most layer, turning the surface hydrophobic (CA ∼87°). Conversely, when the brushes are treated with acidic water, P2VP chains are solvated and able to stretch away from the surface, turning it hydrophilic (CA ∼18°). The chemical composition of these mixed polymer brushes after treatment with different solvents was confirmed by X-ray photoelectron spectroscopy (XPS) as the soluble polymer chains are preferentially enriched at the outermost layer after drying.149
 |
| | Fig. 6 Schematic diagram showing switching behavior of binary polymer brushes upon treatment with selective solvents.148 | |
At small grafting densities (so called “mushroom regime”) the response of grafted chains is very similar to that of the bulk polymer solution.145 At high grafting densities the collapse is weak and the brush forms a homogeneous layer, which is slightly thinner in a poor solvent than in a good solvent or in the θ regime. That is because of the very crowded layer of strongly stretched polymer chains when there is no free space for conformational changes. In the grafting density regime, in which layers vary between very low and high surface coverage regimes, the brushes are unstable in poor solvents and form clusters (pinned micelles) on the surface to avoid unfavorable interactions with the poor solvents.161,162 In a good solvent the brush is swollen and it forms a homogeneous layer of stretched tethered chains. In this grafting density regime (cross-over regime) the polymer brush demonstrates the most pronounced response to solvent quality.163 Thus, we may conclude that the largest conformational response in a polymer brush is in the very beginning of the brush regime when chains start to overlap. Switching a range of the thin polymer film properties relevant to density and thickness of the film can be easily approached with the brushes of moderate grafting densities. It is noteworthy that the optimal grafting density may be affected by a concentration dependency of the x-parameter for some polymer–solvent systems. For example, the high responsiveness (collapse in water above a lower critical solution temperature) of both brush thickness and density profile is sensitive to grafting density and solvent quality. The responsive behavior of the homopolymer brush is referred to changes of free energy of the brush in its environment due to the change of solvent quality. Any application of the responsive homopolymer brushes will be grounded on this principle.
Unlike conformation transition of brushes in solution, solid state conformation changes driven by exposure to different kinds of solvent are particularly interesting due to the possibility of both in situ visualization by AFM and manipulation of the conformational properties of molecular brushes. Gallyamov and co-workers studied conformational transitions of PBPEM-g-PBA brushes induced by cyclic exposure of the wafers with the adsorbed brushes to water and alcohol vapors.164,165 Exposure to saturated alcohol vapor induced collapse of the adsorbed individual polymer chains while exposure to saturated water vapor promoted their extension.166 In the presence of the alcohol layer with the lower surface energy, these brushes tend to minimize the occupied surface area through partial desorption of the PBA side chains leading to a transition from an extended to a globular conformation. Upon condensation of water vapor, PBA side chains re-adsorb to the substrate and cause extension of the backbone.
3.2.2 pH-responsive polymer brushes.
All pH responsive polymers contain pendant acidic (e.g. carboxylic and sulfonic acids) or basic (e.g. ammonium salts) groups that are capable of either accepting or releasing protons in response to environmental changes in pH.167 Changes in the environmental pH thus lead to conformational changes of the soluble polymers and a change in the swelling behavior of the hydrogels when ionizable groups are linked to the polymer structure. Therefore pH is an important signal, which can be addressed through pH-responsive materials.168–174 Ionizable polymers with a pKa between 3 and 10 are candidates for pH-responsive systems. Weak acids and bases like carboxylic acids, phosphoric acid and amines exhibit a change in the ionization state upon variation of the pH. This leads to a conformational change for the soluble polymers when these ionizable groups are linked to the polymer structure. For example, the pKa of poly (acrylic acid) (PAA) depends on molecular weight and ranges within 6.8–7 for molecular weights of order 100 kDa.175 When the pH of the solution is below the pKa value, the polymer is in a compact, collapsed form. As the pH increases above the pKa, the polymer exhibits the fully stretched conformation due to the electrostatic repulsion between the segments.176 Classical monomers are acrylic acid (AA), methacrylic acid (MAA), maleic anhydride (MA), and N,N-dimethylaminoethyl methacrylate (DMAEMA). The unique properties of pH-responsive polymers arise from the facile pH adjustment which induces the ionic interaction and hydrogen bonding, resulting in a reversible microphase separation or self-organization phenomenon. Thus, pH responsive polymeric systems provide the possibility for the preparation of smart functional materials which can be used for potential therapeutic applications, e.g., controlled drug delivery based on pH-triggered release.
A change in the ionization state caused by pH variation usually accompanies conformational changes of single molecular brushes. Atomic force microscopy was used to study the conformation of adsorbed brushes as a function of pH. The adsorbed molecules undergo a globule-to-extended conformational transition as the solution is changed from acidic to basic. This transition was monitored on a mica surface by imaging individual molecules with AFM.177
Poly-2-(dimethylamino)-ethyl methacrylate (PDMAEMA) is a unique responsive polymer since it responds to temperature and also to pH in aqueous solution. It can also be permanently quaternized and converted to zwitterionic structures (via reaction with propanesultone), forming materials with upper critical solution temperature (UCST) properties, as described in the previous section. As expected, the structural changes were induced by variation of pH, ranging from 2 to 10.178 At pH 7, the PDMAEMA brushes formed worm-like structures that can be quite curved. At pH 2, most of the brushes are protonated and ionized, showed more stretched morphologies. More remarkably, at pH 10, the brushes are strongly contracted with an average length around 110 nm, which is attributed to a collapse of the non-ionized PDMAEMA side chains. pH responsive PDMAEMA brushes were also synthesized from a conductive polythiophene (PT) backbone by Winnik et al.179 They observed conformational transitions of PT-g-PDMAEMA with a change in pH that contributed to spectral shifts. As shown schematically in Fig. 7, the polymer brush forms a more extended conformation with a decrease in pH from 8 to 2.
 |
| | Fig. 7 Proposed mechanism for the molecular conformational transition accompanying the change of solvent polarity or the change of pH in water.179 | |
3.2.3 Temperature-responsive polymer brushes.
The effect of temperature on the temperature-responsive copolymer brushes focuses on the point of the critical solution temperature in a special medium. When the temperature of the medium nears to upper or lower the critical solution temperature of the polymer, the brushes will exhibit a sudden change in surface morphology. Such copolymers, which become insoluble upon heating, have a lower critical solution temperature (LCST). Conversely, systems which become soluble upon heating have an UCST. Thermodynamically, the LCST and UCST behavior of polymers can be explained as a balance between the entropic effects of the dissolution and the ordered state of water molecules in the vicinity of the polymer and the enthalpic effects due to hydrogen bonding and hydrophobic interactions. These transitions are observed as coil-to-globule transitions. The physical properties—for example shape, size, color, solubility, viscosity or wettability—of thermoresponsive materials vary in response to mild temperature fluctuations.180 Thus, these types of materials have been shown to be useful for a wide range of applications such as biosensors, implants, scaffolds for tissue engineering or drug-delivery devices, selective bioseparation, phase separation immuno-assays, cell patterning, and DNA sequencing etc.181–191. In particular, PNIPAM has been by far the most studied thermoresponsive polymer in materials science.192
Reversible solubilization of thermoresponsive polymers upon a change in temperature leads to conformational change such as stimulated collapse by dehydration and expansion by rehydration. The resulting conformational changes can be unique for molecular brushes because these temperature driven changes occur on the single molecule level. Water-soluble polymers whose solubility depends on temperature result in materials with a LCST or UCST. While most synthetic water-soluble polymers become more soluble upon heating, some of them precipitate in solution. Polymers with tunable LCST are of increasing interest for biological applications, because they are soluble in aqueous solution below their LCST through hydrogen bonding with water molecules, but become dehydrated and insoluble when heated above the LCST, resulting in abrupt phase separation. For example, thermoresponsive linear polymers such as PNIPAm, exhibit sharp coil–globule transition in water at 32 °C, changing from a hydrophilic state below this temperature to a hydrophobic state above it.193–195 The phase transition, as shown schematically in Fig. 8, and hence the origin of the ‘smart’ behavior, arises from the entropic gain as water molecules associated with the side-chain isopropyl moieties are released into the bulk aqueous phase as the temperature increases past a critical point.
 |
| | Fig. 8 Schematic of ‘smart’ polymer response with temperature.42 | |
An interesting UCST polymer brush, poly [2-(methacryloyloxy)ethyl]-dimethyl(3-sulfopropyl) ammonium hydroxide (PMEDSAH), was synthesized and characterized by Azzaroni et al.196 Zwitterionic PMEDSAH brushes exhibit a complex temperature behavior that depends on the PMEDSAH molecular weight and grafting density, and results in various inter- and intrachain associated states.197 By lowering the surface graft density, the magnitude of the contact angle change could be increased from about 10° (100% initiator grafting density) to about 35° at 10% initiator graft density. Sun et al.198 grafted thermally responsive PNIPAAM brushes on both a flat and a rough silicon substrate via surface initiated atom transfer radical polymerization. However, reversible, thermoresponsive switching between superhydrophilic (∼0°) and superhydropobic (∼150°) states was realized only on the microscopically rough surfaces (Fig. 9a). Similarly, Fu et al.199 realized a dynamic superhydrophobic to superhydrophilic switch by synthesizing a PNIPAAM brush on a nanoporous anodic aluminum oxide surface200 (Fig. 9b).
 |
| | Fig. 9 (a) Switching between superhydrophilicity and superhydrophobicity of a PNIPAAM-modified rough silica surface,212 and (b) representative topographical atomic force microscopy (AFM) images of PNIPAAM grafted of a PNIPAAM-modified nanoporous, structured anodic aluminum oxide membranes in water at 25 °C (left) and at 40 °C (right).199 | |
Another class of thermally responsive systems is based on oligo (ethylene glycol) methyl ether methacrylates (OEGMA).201–204 These thermoresponsive polymers containing short oligo (ethylene glycol) side chains were recently proposed as an attractive alternative to PNIPAM. Poly (ethylene glycol) (PEG) is an uncharged, water-soluble, nontoxic polymer used to prepare biocompatible materials, such as biosensors and drug delivery systems.202,203,205–207 Recently, Lutz203 carried out the copolymerization of di (ethylene glycol) methyl ether methacrylate (MEO2MA) with poly (ethylene glycol) methyl ether methacrylate (PEGMA, MW = 475) by ATRP and demonstrated that the LCST of obtained polymer increased with increasing composition of PEGMA. This concept has been applied to molecular brush systems by Matyjaszewski and co-workers.65 Molecular brushes with statistical or block copolymers of di (ethylene glycol) methyl ether methacrylate (MEO2MA) and tri (ethylene glycol) methyl ether methacrylate (MEO3MA) have been prepared by grafting from a poly (2-(2-bromoisobutyroyloxy)ethyl methacrylate (PBIEMA) macroinitiator via ATRP.208 In the case of brushes with statistical copolymer side chains, the LCST increased with the mole fraction of MEO3MA in the side chain, and the hysteresis between the heating and cooling cycles decreased with the length of the side chain. For brushes with block copolymer side chains, the cloud point of the block brushes solution displayed two stages of aggregation during heating, exhibiting the results of both intermolecular and intramolecular aggregation.
As many other methacrylate monomers, OEGMA can be polymerized under controlled radical polymerization methods yielding well-defined materials. Polymers constructed from these PEG macromonomers exhibit fascinating solution properties in aqueous media. Depending on the molecular structure of their monomer units (i.e., nature of the polymerizable moiety, length of the PEG side chain, end group of the PEG side chain), the polymers can be insoluble in water and readily soluble up to 100 °C or even show thermoresponsive behavior. It is worth noting that, while temperature-responsive behavior of polymers is predominantly monitored in solution, their solid state responsiveness has also been studied by differential scanning calorimeter (DSC) and dynamic mechanical analysis (DMA).209
3.2.4 Mechanically-responsive polymer brushes.
As already discussed, changes in the surrounding environmental conditions can trigger chemical and conformational changes in stimulus-responsive polymer brush layers.210–212 It is, however, also possible for a stimulus-responsive polymer system to change chemically in response to a mechanical stimulus. This is similar to the known phenomenon of mechanotransduction in biology, i.e., the ability of cells to convert a mechanical stimulus into an electrical signal.
Engineering new polymer surfaces involves designing complex architectures with features such as graded branching and composition that will lead to novel material properties in terms of mechanical behavior, adaptability, and functionality. Nanoscale devices and their operating environment require adaptive surfaces constructed with smart properties that can not only sense or respond to environmental stimuli but can also be robust and possess tailored, on-demand physical properties.213–215 The polymer brush layers are chemically tethered to the surface at one end, virtually any chemistry can be designed into the layer depending on intended surface interactions, and the high grafting density combined with uniformity in composition, thickness, and structure allows the entire surface to respond to local environmental stimuli.216 They inherently provide the ability to control and change surface composition, allow on-demand properties, and are becoming increasingly significant for practical application in the fields such as nanoscale lubrication, sensing, and biocompatibility.217–220 Therefore, polymer brushes are considered as the ideal choices in such applications.
Molecular brushes change their shape upon variation of film pressure during either lateral compression50,221–223 or spreading224 on a substrate. Similar to the effect of the vapor pressure discussed above, the transition between extended and globular conformations occurs due to the interplay of energetically favored adsorption and entropically disadvantaged stretching of the PBA side chains. AFM allows in situ tracing of the pressure-responsive macromolecules as they change their shape in response to the pressure gradient during spreading on a solid substrate. Unlike static films, which have the same conformation across the film, molecules in a flowing monolayer are unevenly compressed due to the pressure gradient in the flow direction. This system is particularly interesting since lubrication properties of thin layers in micromechanical joints depend on pressure, which controls the substrate coverage as well as conformation and orientation of the adsorbed molecules. In addition to the backbone extension, steric repulsion between the densely grafted side chains causes significant tension in the backbone covalent bonds. The bond tension depends on molecular architecture and interaction of macromolecules with the surrounding environment.225 The unique property of these miniature tensile machines is that the bond tension is self-generated without applying any external force. One can control the bond tension in a broad range from pico- to nano-Newtons through variation of temperature, solvent quality, and substrate composition. In solution, the highest tension, which is developed in maximally dense brushes in an athermal solvent, can be calculated as f = f0N3/8, where N-degree of polymerization of side chains and f0 = kT/b ≅4 pN. For example, for a molecular brush with N = 100 and monomer length b = 1 nm, the backbone tension ranges from f = f0 = 4 pN in a melt to f = f0N3/8 ≅20 pN in a good (athermal) solvent. This tension may be enough to break a weak hydrogen bond; however, it is too small to rupture a covalent bond. In order to achieve higher tensions, one should change the molecular design. Recently, Panyukov et al.226 have shown that bond tension in the order of 1 nN can be developed in a short space of pom-pom macromolecules.
The kinetics of backbone scission has been studied as a function of substrate surface energy.227 The scission rate revealed extremely high sensitivity to the surface energy of the substrate. Fig. 10 shows kinetics of the decrease of the average polymer chain length (L) on substrates with different surface tension γ. It shows that the rate constant of the scission reaction increases by almost 2 orders of magnitude when the surface tension of the underlying substrate increases by just 5%. Fig. 11 shows a series of AFM images obtained from each of the brushes exposed to aqueous substrates with different surface energies for a time interval of 62 min. One can see that the molecules are noticeably shorter on substrates with a higher surface energy. As such, these polymer architectures can be regarded as miniature tensile testing machines, wherein the brush section generates force of a desired magnitude and transmits it to covalent bonds in the backbone. These molecular devices can be used for mechanical activation of chemical reactions at specific chemical bonds within a macromolecule. For example, brushes with a disulfide (S–S) bond in the middle of the all-carbon polymethacrylate backbone have been synthesized to demonstrate that only one specific bond within a macromolecule can be broken while leaving other bonds intact.228
 |
| | Fig. 10 Kinetics of the decrease of the average polymer chain length (L) on substrates with different surface tension.227 | |
 |
| | Fig. 11 AFM height images of molecular brushes taken up on mica surfaces after spending 62 min on aqueous substrates with different surface tensions.227 | |
3.2.5 Electro- and magnetic-responsive polymer brushes.
Comparing with the single pH-responsive system, an electrical field in the form of an external stimulus offers numerous advantages (e.g. availability of equipment). This form of an external stimulus also allows for precise control over the magnitude of the current, the duration of electrical pulses and the interval between pulses.229 These systems exploiting this external stimulus are prepared from polyelectrolytes, which are polymers that contain a relatively high concentration of ionizable groups along the backbone chain. This property renders these polymers pH-responsive as well as electro-responsive.167,229 Under the influence of an electric field, electro-responsive hydrogels generally shrink or swell and this property has allowed its application in drug delivery systems, artificial muscle or biomimetic actuators.230
Molecular brushes with block copolymer side chains can also act as templates for the preparation of nanostructured hybrid materials by coordination with metal ions. Various metal, metal oxides, and semiconducting nanoparticles have been prepared in this way. Similarly, the cylindrical shape of brushes with diblock copolymer side chains composed of PAA-b-PBA has been used as a single molecular template for the preparation of magnetic nanoparticles, wherein PAA core blocks coordinate with Fe2+ or Fe3+ ions and the PBA shell provides the stability of nanoparticles.53 Temperature-dependent magnetic properties of the hybrid nanocylinders have been investigated. In the temperature range of 25–295 K, the fabricated nanoparticles are superparamagnetic because no hysteresis is observed. The ferrimagnetic nature of the fabricated magnetic nanoparticles, however, was detected at very low temperatures, such as 2 K, showing symmetric hysteresis loop.54
3.2.6 Ion-responsive polymer brushes.
As discussed above, zwitterionic polymers such as poly (sulfobetaine)s have thermoresponsive UCST properties. Another way to achieve the water solubility of poly (sulfobetaine) is to add a simple salt. The site-binding ability of the cation and the anion allows polymer chains preferentially to complex the low molecular weight electrolyte and reduce the attractive inter-chain interaction, leading to chain expansion in aqueous solution. For cationic polyelectrolyte brushes, however, addition of salt was found to screen the electrostatic interactions within the polyelectrolyte, resulting in conformation changes from stretched to collapsed form. Indeed, the quaternized PDMAEMA brushes collapse in solution with high concentration of monovalent salt,178 in the presence of a di- or trivalent salt, this collapse passes through an intermediate state in which the cylindrical polymer brushes assume a helical conformation. The formation of the helix is prompted by the contraction of the polymer molecules along the long axis by the presence of the multivalent ions.
3.2.7 Complex-responsive polymer brushes.
Recently, the research interest of the stimuli-responsive behavior of smart polymers has been extended to multiple external stimuli from single stimuli. Of special importance seem to be polymers that are responsive to light and temperature, and accordingly, there have been several reports on temperature-responsive poly (N-alkylacrylamide) copolymers, for example, those containing light-responsive azobenzene in the side groups of the polymer chain.231–235 Azobenzene groups are known to undergo a reversible isomerization from trans- to cis-configuration upon irradiation.236 In the excited cis-configuration, the higher dipole moment leads to an increase of local polarity of the polymer chain, which causes an increase of the LCST.234–235,237 As a result, these copolymers can be precipitated upon irradiation with UV light within a certain temperature range. The azobenzene chromophores were incorporated as side groups in the polymer backbone and not at the chain end. Upon increasing the number of azobenzene units, the photoinduced shift increased up to a certain value but decreased thereafter. These phenomena might be due to the variety in the number of azobenzene moieties and side-group effects of the photofunctional units. Akiyama and Tamaoki238 demonstrated that photoisomerization of a single terminal unit of a polymer could also trigger a phase transition of a polymer chain. They prepared an end-functionalized PNIPAM by atom transfer radical polymerization with an azobenzene derivative initiator. A linear increase of the LCST shifts with increasing amount of azobenzene located at the end group was found by this group, which was in contrast to the transition behavior of azobenzene-functionalized PNIPAM copolymers.
A growing trend in designing stimuli-responsive polymeric solutions is toward creating systems with multiple-responsive components, with covalently or physically bonded segments responsive to different stimuli. One example of the multi-responsive switchable copolymer is based on temperature responsive PNIPAM and pH- and photo-responsive spirobenzopyran which exhibits temperature, pH, and photo-responsiveness in aqueous solutions. In mixed brushes, two or more different polymers are attached to the same substrate and constitute the brush. Unlike unmixed brush polymer, different polymers in the mixed brush segregate into nanoscopic phases. The phase segregation is a lateral segregation process in a nonselective solvent in which different polymers form spherical or elongated clusters. Both the polymers are exposed on the top of the brush. In selective solvents, the mixed brush structure may be seen as a combination of lateral and layered segregation mechanisms. In the latter case, one polymer preferentially segregates to the top of the brush, while another polymer forms clusters segregated onto the grafting surface. The most important difference of the mixed brush compared to the homopolymer brush is that not only the height and density profile but also the composition profile depends on the solvent quality. In other words, the surface composition of the brush is switched by a change in its environment.
Much like AB diblock copolymers, when molecular weights are sufficiently high, the two immiscible homopolymers in mixed brushes will undergo microphase separation; the covalent fixation of one end of polymer chains on a solid surface prevents macroscopic phase segregation. The phase behavior of binary mixed homopolymer brushes on a planar solid substrate is dictated by a number of factors, including (i) degree of polymerization (DP) of each grafted polymer (NA and NB), (ii) Flory–Huggins interaction parameters between two components (χA–B), between each polymer and environment (χA-E and χB-E), and between each polymer and the grafting substrate (χA–S and χB–S), where A, B, E, and S represent respectively polymer A, polymer B, environment E (E could be a solvent or a polymer matrix, etc.), and substrate, (iii) grafting density of each polymer, and (iv) distribution of grafting sites of two polymers on the substrate.
When AB binary brushes, which consist of hydrophilic and hydrophobic homopolymers, contact with a hydrophilic solvent the hydrophilic constituent segregates to the surface, vice versa. Besides the segregation perpendicular to the surface the laterally structured phase might become stable, but forms dimples, i.e., small clusters on the surface which are separated by a distance of chain extension. If the solvent is bad for one component and good for the other component surface micelles form, i.e., the component with the low solvent affinity collapses into a dense core, which is shielded by other components. Theoretical research showed that for equal amounts of A and B chains grafted to the surface Φ = 1/2 as a function of overlap δ and incompatibility χ, the disordered phase which is neither laterally nor perpendicularly segregated, the layered phase (1D), where the symmetry between A and B is spontaneously broken and the two components segregated perpendicular to the surface, the “ripple” phase, in which the two components segregated laterally into symmetrical cylinders, and two “dimple” phases.
The phase diagrams are shown in Fig. 12. The incompatibility χ at which the “ripple” structure forms increases linearly with δ in the limit of strong overlap δ→0. Upon the increasing incompatibility, a second order transition from the disordered phase to the “ripple” phase to an hexagonal “dimple” phase. Fig. 12(b) is the phase diagram for δ = 1/2 as a function of the composition Φ. At high incompatibilities or extreme compositions the majority component forms a hexagonal array of clusters. At lower incompatibilities or more symmetrical composition, a hexagonal “dimple” phase was found, but the minority component forms the clusters.
 |
| | Fig. 12 (a) Phase diagram for a symmetric polymer brush (Φ = 1/2): under good solvent conditions a transition between a disordered and a “ripple” phase can be found. In bad solvent conditions the disordered, the ripple and two “dimple” phases are stable. (b) Phase diagram for δ = 1/2 as a function of the composition Φ and the incompatibility χ , “dimple A” and “dimple B” denote the hexagonal arrangement of A rich and B rich clusters, respectively. The inset displays the variation of the lateral unit cell size L along the “dimple” “ripple” phase boundary. The solid line corresponds to the ripple phase the dashed line corresponds the dimple phase.157 | |
The complexity of the problem, however, substantially increases when the designs of complex responsive systems that mimic the functions of life systems are considered. Such artificially created systems will be capable of reporting on toxins, diagnosing cancer cells, monitoring important parameters of organs, and targeting the release of drugs. Currently, research is focused on how the interactions with polymer brushes may be precisely tuned and monitored in a controlled environment. This has potential benefits for biomaterials, sensors, microfluidic technologies, adhesive materials, micro-actuators, and so forth.
4 Emerging applications of SPBs
The major objective for the application of SPBs is to regulate, adjust, and switch interaction forces between the brush and its environment constituted of liquid, vapor, solid, another brush, particles, and so forth. The simplest formulation of the problem is switching between attraction and repulsion. For example, the polymer brush-like layer stabilizes colloidal dispersion. However, upon a stimulus (a change of its environment or medium, actually), the colloid coagulates because the repulsive forces of the brush have been “switched off.” This simple effect has numerous important applications in various technologies, and it has not yet been fully explored and engineered. The same simple problem is important if the friction coefficient and adhesion, or wetting could be rapidly changed to switch off and on for capillary flow,189 cell adhesion, protein adsorption,42 cell growth,23 membrane permeability239 and drug release.240 Therefore, polymer brushes with different stimuli-responsive properties may be applied to different fields.
4.1 Micro/nanomaterial system fabrication
The stimulus-response capability of polymer brushes could be used as a triggered “catch and release” surface for nanomaterial fabrication. These brush surfaces provide interesting substrates for making novel devices, the preferential chain orientation of brushes on the surface can also be used to template micro/nanoscale materials. For example, Comrie and Huck210 harnessed this capability for the fabrication of polymer–metal micro-objects. They incorporated a hydrophobic functional group into poly (glycidyl methacrylate) (PGMA) brushes, as a means of creating a robust etch-resistant film on a chromium substrate. As shown in Fig. 13, Tugulu et al.241 used poly(methacrylic acid) (PMAA) brushes as a novel template to form microstructured, crystalline calcite thin films. This strategy makes use of photo-lithographically patterned PMAA brushes grown by SI-ATRP and capitalizes on the film's ionic structure. When supersaturated calcium carbonate solution is passed over the PMAA brush, the brush templates the calcium carbonate through ionic interactions and, upon annealing, produces a crystalline calcite film that is an exact 3D reproduction of the patterned PMAA brush. The use of SPBs in this approach may bestow additional control over the template morphology by inducing compressive or lateral stresses that enable nanofilm porosity control or finely tuned fractal geometries. For structural patterning, it should be become an interesting development in the future to use SPBs and selective deposition techniques.
 |
| | Fig. 13 A novel approach to form microstructured, crystalline calcite thin films. Photo-lithographically patterned poly (methacrylic acid) (PMAA) brushes are grown by SI-ATRP and capitalize on the film's ionic structure, and the preferential chain orientation of brush thin films can be used to template micro/nanoscale for novel device fabrication.241 | |
4.2 Lubrication of surfaces
Polymer brush lubrication mainly stems from the desire for reducing or controlling the friction forces between surfaces in relative sliding motion.139,242–246 While the observation of extremely small kinetic friction coefficients has stimulated first interesting applications, as, for example, in the design of artificial joints,243 the theoretical understanding is still incomplete.247 Since most of the investigations have been devoted to (quasi-) stationary processes, such as for instance steady or oscillatory shear motion, little is known about nonstationary polymer-brush lubrication. The same applies to polymer-brush lubricated surfaces with embedded macromolecules, as they appear in nature, e.g., in mammalian synovial joints.248
Apart from synovial joints, many other biological systems contain brush-like structures and most of them also contain macromolecular inclusions, e.g., capillaries in plants or bloodstream. Such inclusions increase the frictional loss within the bilayer, but bear the distinct advantage of stabilizing nonstationary processes. This should be very important for the understanding of biological transport processes.
4.3 Novel biological sensors
Smart polymer brush films have been designed by using a variety of approaches, including reversible photoisomerisation reactions, reversible swelling/collapsing of grafted polymers, and phase separation in mixed grafted brushes or diblock copolymers.15,249 These surfaces are capable of responding to very subtle changes in the surrounding environment such as light, temperature, salt concentration and pH.250 The macroscopic responses are caused by the reorganization of the internal or external surface structure of the deposited polymer layers. Chemically- and biochemically-responsive surfaces offer intriguing possibilities for the development of novel biological sensors,
4.3.1 Electrochemical DNA (E-DNA) sensors.
Indeed, smart polymer brush surfaces that respond to specific chemical and biological species have been the basis for the fabrication of highly sensitive, reagentless, re-usable biosensors. One recent development is the electrochemical DNA (E-DNA) sensor,251–253 which is the electrochemical equivalent of an optical molecular beacon-oligonucleotide254 probe that becomes fluorescent upon hybridization with target DNA molecules. The detection method in the E-DNA sensor is based on the alteration of the electron transfer dynamics as a consequence of a structural rearrangement induced by target hybridization. An E-DNA sensor is comprised of a surface-confined DNA stem-loop labeled with an electroactive reporter as the hybridization sensing element255 (Fig. 14a). In the absence of a target, the stem-loop holds the redox moiety (e.g. ferrocene, Fc) in proximity to the electrode, producing a large Faradaic current. Other directly related sensors based on binding-induced folding of aptamers have been developed.256,257
 |
| | Fig. 14 (a) Signal-off E-DNA sensor based on a surface-confined stem-loop oligonucleotide that holds the ferrocene (Fc) group into close proximity with the gold electrode surface, thus allowing facile electron transfer from the redox group to the electrode. On hybridization with the target sequence, the distance between the Fc group and the electrode is altered, decreasing the electron transfer efficiency,225 (b) and (c) signal-on E-DNA sensors, wherein a large detection signal arises upon hybridization with target DNA.253,256 | |
4.3.2 Electrochemical aptamer-based (E-AB) sensors and cell-based sensors.
E-AB sensors and cell-based sensors based on binding-induced folding of aptamers have been developed.257 Aptamers are DNA or RNA sequences selected in vitro from combinatorial libraries by systematic evolution of ligands by exponential enrichment (SELEX). Aptamers can be selected against diverse targets, such as dyes, proteins, peptides, aromatic small molecules, antibiotics and other biomolecules, with high specificity and affinity,258 and thus they are particularly useful as the basic sensing element for biosensor applications. A series of novel electrochemical aptamer-based (E-AB) sensors (Fig. 14b, c), an analogous version to the E-DNA sensor, have been reported for such diverse targets as the blood-clotting enzyme thrombin,256,257 the small molecule cocaine259 and adenosine triphosphate (ATP).260 Cell-based sensors have also been exploited. An elegant approach to establish molecular communication between cells and material surfaces based on enzyme-responsive SAMs was introduced by Mrksich et al.261 The authors demonstrated262 first that 4-hydroxyphenyl valerate-terminated SAMs on gold electrodes could be switched enzymatically by cutinase from a redox inactive molecular brush surface to a redox active molecular brush surface.
4.4 Drug delivery
Conventional drug delivery methods physically entrap molecules within a polymer lattice; the drug is released slowly by diffusion or upon degradation of the network. These methods typically result in an early peak in plasma drug concentration followed by a steady, linear release. This is far from ideal because the local drug concentration and location of delivery is not precisely controlled. Below the therapeutic dose, the drug is ineffective whereas high concentrations of drug may be toxic or lead to undesirable side effects. Polymers have been used to tailor drug release, which maintains the drug concentration within the desired therapeutic range. However, such controlled release systems are insensitive to metabolic changes in the body and are able to neither modulate drug release nor target the drug to diseased tissue. This lack of control has motivated the exploitation of bioresponsive polymers as drug carriers. More recently, drug carriers that respond to magnetic fields,263 light,264 radiation,265 and ultrasound240 have also been developed. These external stimuli allow for greater control over when and where the drug is released. By tuning the formulation or chemical moieties of the polymer, the sensitivity to the stimuli can be precisely controlled.
Externally regulated drug delivery systems allow for pulsatile drug delivery, which may be defined as the rapid and transient release of a certain amount of drug within a short time period immediately after a predetermined off-release period.266 Targeted drug delivery systems allow for drug delivery to specific sites, organs, tissues or cells in the body where drug therapy is required, e.g. the specific targeting of drugs to cancer cells.267 A very effective way of achieving site-specific drug targeting is by employing stimuli-responsive polymers. In the field of drug delivery it is the LCST of polymers and systems that is generally more relevant. In terms of thermodynamics, the phenomenon of polymer aggregation at the LCST is owed to the entropy (S) of the two-phase polymer and water system.268 A positive ΔS allows the increase in temperature to contribute to the system aggregation. This is also encouraged by the positive enthalpy ΔH (which is smaller than the entropy term). Under these circumstances, association is favorable as the free energy of association (ΔG = ΔH − TΔS) is negative.269 Theoretically, a negative excess entropy of mixing is required for an LCST; however, the nature of the polymer–water interactions may also be responsible.193,270–274
Electroactive functionalized surfaces based on the hydroquinone–quinine redox couple have been shown to give real-time control over the molecular interactions that mediate peptide attachment (Fig. 15). The electroactive monolayers were able to directly switch peptide ligand activities on and off, and subsequently to influence the behavior of attached cells in situ and in real time.275 This dynamic property was based on the use of the electroactive O-silyl hydroquinone moiety to tether the arginine–glycine–aspartic acid (RGD) peptide ligand to the monolayer. Upon electrochemical oxidation of the O-silyl hydroquinone to the corresponding benzoquinone moiety, the silyl ether was hydrolyzed and the RGD peptides were selectively released from the surface.
 |
| | Fig. 15 Selective release of the RGD peptide from a monolayer presenting the O-silyl hydroquinone by electrochemical oxidation and subsequent immobilisation of a second RGD peptide by a Diels–Alder reaction.262 | |
4.5 Electrically-controlled switchable biological surfaces
The molecular brush surfaces with a number of different electroactive groups have been successfully employed to switch on functionalities in situ, offering an unprecedented ability to manipulate the interactions of DNA, proteins, and cells with brush surfaces.262 Electrically conducting polymers are of considerable interest for a variety of biomedical applications.276 Conducting polymer is generally comprised of a conjugated polymer chain with π electrons delocalised along the backbone, yielding a semiconducting polymer that can be reversibly tuned through doping, an oxidation–reduction process where charge carriers are introduced to the polymeric backbone either chemically or electrochemically.
Polypyrrole is a particularly interesting candidate for biomedical applications, by virtue of its chemical and thermal stability, and low cytotoxicity. Apart from being exploited in drug-delivery systems,277,278 polypyrrole polymers may be especially useful as advanced substrates for cell cultures since they provide a non-invasive way to regulate cell form and function (e.g. DNA synthesis). By reversibly changing their oxidation state and, consequently, their properties and surface binding characteristics, polypyrrole polymer films on indium tin oxide (ITO) coated glass substrates have been shown to act as dynamic cell culture substrates.279In vitro studies demonstrated that extracellular matrix molecules, such as fibronectin, adsorb efficiently onto the oxidised (polycation) polypyrrole thin films, and support cell attachment under serum-free conditions.
Low density polymer brushes on gold have attracted interest for controlling protein adsorption and release under electrical modulation.280,281 These surfaces display increased interchain distances, enabling the reversible conformational transition of surface-confined molecules. Amino-terminated monolayer polymer brushes induced a neutral and hydrophobic surface under a negative potential and a positively charged and hydrophilic surface under a positive potential. These low density SAMs have been successfully integrated in microfluid chips to reversibly control the assembly of two proteins with different isoelectric points281 (Fig. 16).
 |
| | Fig. 16 Amino-terminated monolayer brushes show reversible and different conformational reorientation behavior under negative and positive potentials.281 | |
4.6 Thermo-responsive culture dishes for cells
A class of thermally-responsive polymer surfaces that regulate molecular recognition events and control cell attachment and detachment without cell damage has been studied during the past few years. The most widely studied system is PNIPAM, which has a LCST of 32 °C in aqueous solution. Below its LCST, PNIPAM polymer is in an extended, solvent-swelled conformation, but when heated up above the LCST, the polymer undergoes a phase transition to yield a collapsed morphology that excludes solvent. This behavior is based on a widespread hydrogen bond network between the amide groups and water molecules at lower temperatures, whereas at higher temperatures the stabilizing H-bonds break up and the hydrophobic interactions become predominant222 (Fig. 17).The fact that PNIPAM undergoes a sharp property change in response to a moderate thermal stimulus near physiological temperatures has been utilized to develop thermo-responsive culture dishes for cells.
 |
| | Fig. 17 Diagram illustrating the temperature-induced switching of a PNIPAM-modified surface.298 | |
4.7 Permeability switching in micro/nanoporous membranes
The possibility of dynamic control of the permeation of chemicals through nanoporous membrances26,282,283 or the interaction of molecules and ions with a responsive surface284,285 offers a unique opportunity for bioseparation.286,287 Surface-grafted SPBs provide an exciting means for controlling permeation through nano- and microporous membranes. The application of smart polymer surface to filtration systems can possibly improve filter life, streamline cleaning cycles, or provide antimicrobial properties to the surface. An advancement of this, however, can be envisioned with the application of SPBs within a filter pore, providing mechanical control over head loss, permeation flow rates, or particle size distribution.288–290 These pore-mediated transport structures include polymer micro/nanoporous membranes,291,292 porous silica materials,293 carbon fibers,294 porous alumina membranes,295 with potential applications in separation science.239
Lokuge et al.239 reported that grafting SPBs to membrane surfaces is an effective route to obtaining controllable, active filtration capabilities. Their nanocapillary arrays, made of Au-coated track-etched polycarbonate, consisted of 80–200 nm regularly arranged pores (Fig. 18). In their approach, physical changes in a surface-grafted PNIPAAM film can be triggered by heating the polymer above its LCST; this causes the membrane permeability for dextran molecules to be switched on because the effective membrane pore diameter increases with decreasing film swelling above the LCST. By tuning the molecular weight and graft density so that brush height is on the order of the pore diameter, control over hydraulic permeability can thus be exerted.
 |
| | Fig. 18 Reversible switching capability of a PNIPAAM-grafted NCAM membrane. Permeation of 77 kDa dextran through an ID200/Au50/PNIPAAm 10 nanocapillary array membrane (NCAM) over several heating–cooling cycles.239 | |
4.8 Tunable catalysts
The possibility of exposing or hiding functional group or nanoparticles in the reconstructable surfaces has opened new directions in chemical and biochemical catalysis. These surfaces can be created by attaching some smart polymers. Since the polymer brushes dictate how hairy particles interact with environment, the conformation changes of individual polymer chains could cause the particles to exhibit different behavior. A representative example of such polymers is thermosensitive hydrophilic polymers that can undergo coil-to-globule or hydration-to-dehydration transitions in water at lower critical solution temperatures (LCSTs). Just like what is shown in Fig. 19, temperature-dependent swelling and shrinking of shell were used to alternatively expose and hide the silver nanoparticles on the surface of the colloids. The reagents at low temperature can diffuse freely to the nanoparticles that act as catalysts. However, the network shrinks and the catalytic activity of the nanoparticles is strongly diminished at higher temperatures (T > 30 °C). Conjugation of stimuli-responsive polymeric systems with catalytic nanoparticles and enzymes could thus create new opportunities for bio- and chemical technologies.296
 |
| | Fig. 19 PS-NIPA-Ag composite particles consisting of thermosensitive core–shell particles in which Ag nanoparticles are embedded.296 | |
5 Conclusion and outlook
SPBs can be used for a variety of applications, such as switching surfaces and adhesive, protective coatings that adapt to the environment, artificial muscles, sensors and controllable drug delivery. The field of SPBs is a continuously expanding area of research. The expansion is not very fast because of the complexity of the systems for the fabrication as well as for investigations.
Although nature serves as the model for smart polymers and provides many inspirations for designing and developing new materials, making synthetic systems capable of responding to stimuli in a controllable and predictable manner represents significant challenges to create materials that interact with, or respond to, biological environments, especially in mimicking biological systems where structural and compositional gradients at various length scales are necessary for precise and orderly responsive behavior.
There remain, however, challenges ahead in developing more sophisticated stimuli-responsive systems. For example, how might we reach the complexity of stimuli-response that we observe in nature? This goal requires engineered materials with features such as reversible stimuli-response, so that the response can be turned on and off in a controlled fashion. In addition, some particular applications, such as materials for drug release, may require a graded response depending on the intensity of the stimulus. Another challenge that is inherent to almost all organic systems pertains to long-term stability (to temperature, ultraviolet light, solvent vapors) and durability (such as mechanical stability, abrasion). Finally, is it possible to create a stimuli-responsive system wherein a single stimulus produces a cascade of responses in a manner similar to biological systems? The search for smart materials challenges researchers to create fast response, increased sensitivity and robust behavior smart polymers at the molecular level to address our future needs.
Nomenclature
| AFM | atomic force microscopy |
| ATP | adenosine triphosphate |
| ATRP | atomic transfer radical polymerization |
| CMC | critical micelle concentration |
| CRP | controlled/living radical polymerization |
| DMA | dynamic mechanical analysis |
| DMAEMA |
N,N-dimethylaminoethyl methacrylate |
| DP | degree of polymerization |
| LCST | lower critical solution temperature |
| MEO2MA | di(ethylene glyene glycol) methyl ethermethacrylate |
| MEO3MA | tri(ethylene glyene glycol) methyl ether methacrylate |
| MWD | molecular weight distribution |
| OEG | oligo (ethylene glycol) |
| OEGMA | oligo [(ethylene glycol) methacrylate] |
| PBA-b- PtBA | poly (butyl acrylate)-block-poly (tert-butyl acrylate) |
| PBIEMA | poly-2-bromoisobutyloxyethyl methacrylate |
| PEG | poly (ethylene glycol) |
| PEGMA | poly [(ethylene glycol) methacrylate] |
| PGMA | poly (glycidyl methacrylate) |
| PHEMA | poly (2-hydroxyethyl methacrylate) |
| PMAA | poly (methacrylic acid) |
| PMEDSAH | poly [2-(methacryloyloxy)ethyl dimethyl-(3-sulfopropyl)ammonium hydroxide] |
| PNIPAM | poly (n-isopropylacrylamide) |
| P2VP | poly (2-vinylpyridine) |
| PS-b-PtBA | polystyrene-block-poly(tert-butyl acrylate) |
| PtBA-b-PS | poly (tert-butyl acrylate)-block-polystyrene |
| PtBA-b-PBA | poly (tert-butyl acrylate)-block-poly (butyl acrylate) |
| PT-g- PDMAEMA | poly (thiophene)-graft-poly(N,N-dimethylaminoethyl methacrylate) |
| RAFT | reversible addition fragmentation chain transfer |
| RGD | arginine–glycine–aspartic |
| SIP | surface-initiated polymerization |
| SAMs | self-assembled monolayers |
| SPBs | smart polymer brushes |
| SELEX | systematic evolution of ligands by experimental enrichment |
| SFRP | stable free radical polymerization |
| UCST | upper critical solution temperature |
| XPS | X-ray photoelectron spectroscopy |
Acknowledgements
The authors would like to thank Dr Manuel Palacio for reading the manuscript.
References
- S. T. Milner, “Polymer Brushes”, Science, 1991, 251, 905–914 CAS.
- A. Halperin, M. Tirrell and T. P. Lodge, “Tethered chains in polymer microstructures”, Adv. Polym. Sci., 100, 31–71 CrossRef CAS.
- B. Zhao and W. J. Brittain, “Polymer brushes: surface-immobilized macromolecules”, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- S. T. Milner, T. A. Witten and M. E. Cates, “Theory of the grafted polymer brush”, Macromolecules, 1988, 21, 2610–2619 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. K. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, “Emerging applications of stimuli-responsive polymer materials”, Nat. Mater., 2010, 9, 101–113 CrossRef.
- J. Lahann, S. Mitragotri, T.-N. Tran, H. Kaido, J. Sundaram, I. S. Choi, S. Hoffer, G. A. Somorjai and R. Langer, “A Reversibly Switching Surface”, Science, 2003, 299, 371–373 CrossRef CAS.
-
A. M. Urban and M. W. Urban, Stimuli-Responsive Macromolecules and Polymeric Coatings, Oxford University Press and American Chemical Society, Washington, DC, 2005 Search PubMed.
- M. H. Li and P. Keller, “Stimuli-responsive polymer vesicles”, Soft Matter, 2009, 5, 927–937 RSC.
- F. Meng, Z. Zhong and J. Feijen, “Stimuli-responsive polymersomes for programmed drug delivery”, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
-
I. Y. Galaev and B. Mattiasson, Smart Polymers. Applications in Biotechnology and Biomedicine, 2nd edition, CRC Press, Taylor & Francis Group, Boca Raton, 2008 Search PubMed.
- A. Kumar, A. Srivastava, I. Y. Galaev and B. Mattiasson, “Smart polymers: physical forms and bioengineering applications”, Prog. Polym. Sci., 2007, 32, 1205–1237 CrossRef CAS.
- A. S. Hoffman, “The origins and evolution of ‘controlled’ drug delivery systems”, J. Controlled Release, 2008, 132, 153–163 CrossRef CAS.
- D. Tranchida, E. Sperotto, T. Staedler, X. Jiang and H. Schönherr, “Nanomechanical Properties of Oligo(ethylene glycol methacrylate) Polymer Brush-Based Biointerfaces”, Adv. Eng. Mater., 2011, 13, B369–B376 CrossRef.
- Y. Liu, V. Kelp, B. Zdyrko and I. Luzinov, “Synthesis of high-density polymer layers with thickness and grafting density gradients”, Langmuir, 2005, 21, 11806–11813 CrossRef CAS.
- C. Alexander and K. M. Shakesheff, “Responsive Polymers at the Biology/Materials Science Interface”, Adv. Mater., 2006, 18, 3321–3328 CrossRef CAS.
- I. S. Choi and Y. S. Chi, “Surface Reactions on Demand: Electrochemical Control of SAM-Based Reactions”, Angew. Chem., Int. Ed., 2006, 45, 4894–4897 CrossRef CAS.
- W. Senaratne, L. Andruzzi and C. K. Ober, “Self-Assembled Monolayers and Polymer Brushes in Biotechnology: Current Applications and Future Perspectives”, Biomacromolecules, 2005, 6, 2427–2448 CrossRef CAS.
-
B. Bhushan, Springer Handbook of Nanotechnology, Springer-Verlag, 3rd edn, 2010 Search PubMed.
- S. J. Jhaver, M. R. Hynd, N. Dowell-Mesfin, J. N. Turner, W. Shain and C. K. Ober, “Release of nerve growth factor from HEMA hydrogel-coated substrates and its effect on the differentiation of neural cells”, Biomacromolecules, 2009, 10, 174–183 CrossRef.
- S. L. Gras, T. Mahmud, G. Rosengarten, A. Mitchell and K. Kalantar-Zadeh, “Intelligent Control of Surface Hydrophobicity,”, ChemPhysChem, 2007, 8, 2036–2050 CrossRef CAS.
- J. A. Howarter and J. P. Youngblood, “Self-Cleaning and Anti-Fog Surfaces via Stimuli-Responsive Polymer Brushes”, Adv. Mater., 2007, 19, 3838–3843 CrossRef CAS.
- I. Luzinov, S. Minko and V. V. Tsukruk, “Responsive brush layers: from tailored gradients to reversibly assembled nanoparticles”, Soft Matter, 2008, 4, 714–725 RSC.
- P. M. Mendes, “Stimuli-Responsive Surfaces for Bio-Applications”, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- Z. S. Liu and P. Calvert, “Multilayer hydrogels as muscle-like actuators”, Adv. Mater., 2000, 12, 288–291 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. van Duyn, “Biosensing with plasmonic nanosensors”, Nat. Mater., 2008, 7, 442–453 CrossRef CAS.
- I. Tokarev and S. Minko, “Stimuli-responsive hydrogel thin films”, Soft Matter, 2009, 5, 511–524 RSC.
- A. Bhattacharya and B. N. Misra, “Grafting: a versatile means to modify polymers. Techniques, factors and applications”, Prog. Polym. Sci., 2004, 29, 767–814 CrossRef CAS.
- K. Ishizu, “Architecture of multi-component copolymer brushes: synthesis, solution properties and application for nanodevices”, Polym. J., 2004, 36, 775–792 CrossRef CAS.
- M. Wintermantel, M. Gerle, K. Fischer, M. Schmidt and I. Wataoka, “Molecular bottlebrushes”, Macromolecules, 1996, 29, 978–983 CrossRef CAS.
- S. Kawaguchi, K. Akaike, Z. M. Zhang, H. Matsumoto and K. Ito, “Water soluble bottlebrushes”, Polym. J., 1998, 30, 1004–1007 CrossRef CAS.
- S. Lecommandoux, F. Checot, R. Borsali, M. Schappacher and A. Deffieux, “Effect of dense grafting on the backbone conformation of bottlebrush polymers: determination of the persistence length in solution”, Macromolecules, 2002, 35, 8878–8881 CrossRef CAS.
- L. Feuz, F. A. M. Leermakers, M. Textor and O. Borisov, “Bending rigidity and induced persistence length of molecular bottle brushes: a self-consistent-field theory”, Macromolecules, 2005, 38, 8891–8901 CrossRef CAS.
- S. Elli, F. Ganazzoli, E. G. Timoshenko, Y. A. Kuznetsov and R. Connolly, “Size and persistence length of molecular bottle-brushes by Monte Carlo simulations”, J. Chem. Phys., 2004, 120, 6257–6267 CrossRef CAS.
- I. I. Potemkin, A. R. Khokhlov and P. Reineker, “Stiffness and conformations of molecular bottle-brushes strongly absorbed on a flat surface”, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 4, 93–101 CrossRef CAS.
- M. Saariaho, O. Ikkala, I. Szleifer, I. Erukhimovich and G. Ten Brinke, “On lyotropic behavior of molecular bottle-brushes: aMonte Carlo computer simulation study”, J. Chem. Phys., 1997, 107, 3267–3276 CrossRef CAS.
- M. Saariaho, I. Szleifer, O. Ikkala and G. Ten Brinke, “Extended conformations of isolated molecular bottle-brushes. Influence of side-chain topology”, Macromol. Theory Simul., 1998, 7, 211–216 CrossRef CAS.
- M. Saariaho, A. Subbotin, I. Szleifer, O. Ikkala and G. Ten Brinke, “Effect of side chain rigidity on the elasticity of comb copolymer cylindrical brushes: a Monte Carlo simulation study”, Macromolecules, 1999, 32, 4439–4443 CrossRef CAS.
- M. Saariaho, A. Subbotin, O. Ikkala and G. Ten Brinke, “Comb copolymer cylindrical brushes containing semiflexible side chains. A Monte Carlo study”, Macromol. Rapid Commun., 2000, 21, 110–115 CrossRef CAS.
- A. Subbotin, M. Saariaho, O. Ikkala and G. Ten Brinke, “Elasticity of comb copolymer cylindrical brushes”, Macromolecules, 2000, 33, 3447–3452 CrossRef CAS.
- A. Subbotin, M. Saariaho, R. Stepanyan, O. Ikkala and G. Ten Brinke, “Cylindrical brushes of comb copolymer molecules containing rigid side chains”, Macromolecules, 2000, 33, 6168–6173 CrossRef CAS.
- D. Vlassopoulos, G. Fytas, B. Loppinet, F. Isel and P. Lutz, “Polymacromonomers: structure and dynamics in non-dilute solutions, melts, and mixtures”, Macromolecules, 2000, 33, 5960–5969 CrossRef CAS.
- CdLH. Alarcon, S. Pennadam and C. Alexander, “Stimuli responsive polymers for biomedical applications”, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- A. S. Hoffman and P. S. Stayton, “Conjugates of stimuli-responsive polymers and proteins”, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- A. Nelson, “Stimuli-responsive polymers: engineering interactions”, Nat. Mater., 2008, 7, 523–525 CrossRef CAS.
- A. Pelah, A. Bharde and T. M. Jovin, “Protein manipulation by stimuliresponsive polymers encapsulated in erythrocyte ghosts”, Soft Matter, 2009, 5, 1006–1010 RSC.
- F. M. Winnik, D. G. Whitten, M. W. Urban and G. Lopez, “Stimuli-responsive materials: polymers, colloids, and multicomponent systems”, Langmuir, 2007, 23, 1–2 CrossRef CAS.
- Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stoever, “Thermal response of narrowdisperse poly (N-isopropylacrylamide) prepared by atom transfer radical polymerization”, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- H. G. Boerner, D. Duran, K. Matyjaszewski, M. da Silva and S. S. Sheiko, “Synthesis of molecular brushes with gradient in grafting density by atom transfer polymerization”, Macromolecules, 2002, 35, 3387–3394 CrossRef CAS.
- H. Lee, K. Matyjaszewski, S. Yu and S. S. Sheiko, “Molecular brushes with spontaneous gradient by atom transfer radical polymerization”, Macromolecules, 2005, 38, 8264–8271 CrossRef CAS.
- S. J. Lord, S. S. Sheiko, I. LaRue, H.-I. Lee and K. Matyjaszewski, “Tadpole conformation of gradient polymer brushes”, Macromolecules, 2004, 37, 4235–4240 CrossRef CAS.
- N. Khelfallah, N. Gunari, K. Fischer, G. Gkogkas, N. Hadjichristidis and M. Schmidt, “Micelles formed by cylindrical brush-coil block copolymers”, Macromol. Rapid Commun., 2005, 26, 1693–1697 CrossRef CAS.
- M. W. Neiser, S. Muth, U. Kolb, J. R. Harris and J. Okuda, “Micelle formation from amphiphilic ‘cylindrical brush’-coil block copolymers prepared by metallocene catalysis”, Angew. Chem., Int. Ed., 2004, 43, 3192–3195 CrossRef CAS.
- M. Zhang, C. Estournes, W. Bietsch and A. H. E. Mueller, “Superparamagnetic hybrid nanocylinders”, Adv. Funct. Mater., 2004, 14, 871–882 CrossRef CAS.
- M. Zhang, P. Teissier, M. Krekhova, V. Cabuil and A. H. E. Mueller, “Polychelates of amphiphilic cylindrical core-shell polymer brushes with iron cations”, Prog. Coll. Polym. Sci., 2004, 126, 35–39 CAS.
- A. Zhang, J. Barner, I. Goessl, J. P. Rabe and A. D. Schuleter, “A covalentchemistry approach to giant macromolecules and their wetting behavior on solid substrates”, Angew. Chem., Int. Ed., 2004, 43, 5185–5188 CrossRef CAS.
- H. G. Boerner, K. Beers, K. Matyjaszewski, S. S. Sheiko and M. Moeller, “Synthesis of molecular brushes with block copolymer side chains using atom transfer radical polymerization”, Macromolecules, 2001, 34, 4375–4383 CrossRef CAS.
- H. Lee, W. Jakubowski, K. Matyjaszewski, S. Yu and S. S. Sheiko, “Cylindrical core-shell brushes prepared by a combination of ROP and ATRP”, Macromolecules, 2006, 39, 4983–4989 CrossRef CAS.
- H. Lee, J. Pietrasik and K. Matyjaszewski, “Phototunable temperatureresponsive molecular brushes prepared by ATRP,”, Macromolecules, 2006, 39, 3914–3920 CrossRef CAS.
- K. Ishizu and H. Kakinuma, “Synthesis of nanocylinders consisting of graft block copolymers by the photo-induced ATRP technique,”, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 63–70 CrossRef CAS.
- J. Pietrasik, B. S. Sumerlin, R. Y. Lee and K. Matyjaszewski, “Solution behavior of temperature-responsive molecular brushes prepared by ATRP”, Macromol. Chem. Phys., 2007, 208, 30–36 CrossRef CAS.
- D. Neugebauer, Y. Zhang, T. Pakula and K. Matyjaszewski, “Heterografted PEO-PnBA brush copolymers”, Polymer, 2003, 44, 6863–6871 CrossRef CAS.
- D. Wu, Y. Yang, X. Cheng, L. Liu, J. Tian and H. Zhao, “Mixed molecular brushes with PLLA and PS side chains prepared by AGET ATRP and ring-opening polymerization”, Macromolecules, 2006, 39, 7513–7519 CrossRef CAS.
- K. Ishizu, J. Satoh and A. Sogabe, “Architecture and solution properties of AB-type brush-block-brush amphiphilic copolymers via ATRP techniques”, J. Colloid Interface Sci., 2004, 274, 472–479 CrossRef CAS.
- H. Lee, K. Matyjaszewski, S. Yu and S. S. Sheiko, “Hetero-grafted block brushes with PCL and PBA side chains”, Macromolecules, 2008, 41, 6073–6080 CrossRef CAS.
- K. Matyjaszewski, S. Qin, J. R. Boyce, D. Shirvanyants and S. S. Sheiko, “Effect of Initiation conditions on the uniformity of three-arm star molecular brushes”, Macromolecules, 2003, 36, 1843–1849 CrossRef CAS.
- J. R. Boyce, D. Shirvanyants, S. S. Sheiko, D. A. Ivano, S. Qin, H. Boerner and K. Matyjaszewski, “Multiarm molecular brushes: effect of the number of arms on the molecular weight polydispersity and surface ordering”, Langmuir, 2004, 20, 6005–6011 CrossRef CAS.
- M. Schappacher and A. Deffieux, “From combs to comb-g-comb centipedes”, Macromolecules, 2005, 38, 7209–7213 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, “Controlled “living” radical polymerization. Atom transfer radical polymerization in the presence of transition-metal complexes”, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- T. E. Patten, J. Xia, T. Abernathy and K. Matyjaszewski, “Polymers with very low polydispersities from atom transfer radical polymerization”, Science, 1996, 272, 866–868 CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, “Metal-catalyzed living radical polymerization”, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS.
- K. Matyjaszewski and J. Xia, “Atom transfer radical polymerization”, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, “Green atom transfer radical polymerization: from process design to preparation of well-defined environmentally friendly polymeric materials”, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, “Nanostructured functional materials prepared by atom transfer radical polymerization”, Nat. Chem., 2009, 1, 276–288 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, “Narrow molecular eight resins by a free-radical polymerization process”, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, “New polymer synthesis by nitroxide mediated living radical polymerizations”, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, “Living free-radical polymerization by reversible additionfragmentation chain transfer: the RAFT process”, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, “Exploring actuation and mechanotransduction properties of polymer brushes”, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, “Living radical polymerization by the RAFT process”, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- A. P. Vogt, S. R. Gondi and B. S. Sumerlin, “Hyperbranched polymers via RAFT copolymerization of an acryloyl trithiocarbonate”, Aust. J. Chem., 2007, 60, 396–399 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, “Reversible addition-fragmentation chain transfer (RAFT) radical polymerization and the synthesis of watersoluble (co)polymers under homogeneous conditions in organic and aqueous media”, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Moeller, “The synthesis of densely grafted copolymers by atom transfer radical polymerization”, Macromolecules, 1998, 31, 9413–9415 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, “Functional polymers by atom transfer radical polymerization”, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
-
K. Matyjaszewski and T. P. Davis, Handbook of radical polymerization, Wiley, Hoboken, 2002 Search PubMed.
- W. A. Braunecker and K. Matyjaszewski, “Controlled/living radical polymerization: features, developments, and perspectives”, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. A. Davis and K. Matyjaszewski, “Statistical, gradient, block, and graft copolymers by controlled/living radical polymerizations”, Adv. Polym. Sci., 2002, 159, 2–166 CrossRef CAS.
- A. Goto and T. Fukuda, “Kinetics of living radical polymerization”, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- M. F. Cunningham, “Controlled/living radical polymerization in aqueous dispersed systems”, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS.
- M. Wintermantel, M. Schmidt, Y. Tsukahara, K. Kajiwara and S. Kohjiya, “Rodlike combs”, Macromol. Rapid Commun., 1994, 15, 279–284 CrossRef CAS.
- M. Wintermantel, K. Fischer, M. Gerle, R. Ries, M. Schmidt, K. Kajiwara, H. Urkawa and I. Wataoka, “Lyotropic phases formed by molecular bottle brushes”, Angew. Chem., Int. Ed. Engl., 1995, 34, 1472–1474 CrossRef CAS.
- Y. Tsukahara, K. Tsutsumi, Y. Yamashita and S. Shimada, “Radical polymerization behavior of macromonomers. 2. Comparison of styrene macromonomers having amethacryloyl end group and a vinylbenzyl end group”, Macromolecules, 1990, 23, 5201–5208 CrossRef CAS.
- Y. Tsukahara, S. Kohjiya, K. Tsutsumi and Y. Okamoto, “On the intrinsic viscosity of poly(macromonomer)s”, Macromolecules, 1994, 27, 1662–1664 CrossRef CAS.
- M. Gerle, K. Fischer, S. Roos, A. H. E. Mueller and M. Schmidt, “Main chain conformation and anomalous elution behavior of cylindrical brushes as revealed by GPC/MALLS, light scattering, and SFM,”, Macromolecules, 1999, 32, 2629–2637 CrossRef CAS.
- K. Hatada, T. Kitayama, E. Masuda and M. Kamachi, “Electron paramagnetic resonance observation of propagating radicals in the polymerization of highly syndiotactic w-(p-vinylbenzyl)poly(methyl methacrylate) macromonomer”, Makromol. Chem. Rapid Commun., 1990, 11, 101–107 CrossRef CAS.
- E. Masuda, S. Kishiro, T. Kitayama and K. Hatada, “Radical polymerization of highly isotactic and syndiotactic poly(methyl methacrylate) macromonomers”, Polym. J., 1991, 23, 847–857 CrossRef CAS.
- G. C. Berry, S. Kahle, S. Ohno, K. Matyjaszewski and T. Pakula, “Viscoelastic and dielectric studies on comb- and brush-shaped poly(n-butyl acrylate)”, Polymer, 2008, 49, 3533–3540 CrossRef CAS.
- S. Ohno and K. Matyjaszewski, “Controlling grafting density and side chain length in poly(n-butyl acrylate) by ATRP copolymerization of macromonomers”, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5454–5467 CrossRef CAS.
- S. S. Sheiko, M. Gerle, K. Fischer, M. Schmidt and M. Moeller, “Wormlike polystyrene brushes in thin films”, Langmuir, 1997, 13, 5368–5372 CrossRef CAS.
- K. Ishizu, S. Yukimasa and R. Saito, “Organized-polymerization in micelle formed by diblock macromonomers”, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3073–3080 CrossRef CAS.
- K. Ishizu, S. Tsubaki and S. Uchida, “Free-radical polymerization of macromonomers”, J. Macromol. Sci., Pure Appl. Chem., 1995, A32, 1227–1234 CrossRef CAS.
- K. Ishizu, K. Tsubaki and T. Ono, “Synthesis and dilute solution properties of poly(diblock macromonomer)s”, Polymer, 1998, 39, 2935–2939 CrossRef CAS.
- K. Ishizu and T. Furukawa, “Synthesis of functionalized poly(ethylene oxide) macromonomers”, Polymer, 2001, 42, 7233–7236 CrossRef CAS.
- B. Grassl, S. Rempp and J. C. Galin, “New super-hairy semi-rigid polymers”, Macromol. Chem. Phys., 1998, 199, 239–246 CrossRef CAS.
- P. Dziezok, S. S. Sheiko, K. Fischer, M. Schmidt and M. Moller, “Cylindrical molecular brushes”, Angew. Chem., Int. Ed., 1997, 36, 2812–2815 CrossRef CAS.
- E. Nomura, K. Ito, A. Kajiwara and M. Kamachi, “Radical polymerization kinetics of poly(ethylene oxide) macromonomers”, Macromolecules, 1997, 30, 2811–2817 CrossRef CAS.
- K. Ito, K. Tanaka, H. Tanaka, G. Imai, S. Kawaguchi and S. Itsuno, “Poly(ethylene oxide) macromonomers. 7. Micellar polymerization in water”, Macromolecules, 1991, 24, 2348–2354 CrossRef CAS.
- D. Pantazis, I. Chalari and N. Hadjichristidis, “Anionic polymerization of styrenic macromonomers”, Macromolecules, 2003, 36, 3783–3785 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, “An efficient route to macromonomers via ATRP and click chemistry”, Macromolecules, 2006, 39, 5286–5292 CrossRef CAS.
- K. Yamada, M. Miyazaki, K. Ohno, T. Fukuda and M. Minoda, “Atom transfer radical polymerization of poly(vinyl ether) macromonomers”, Macromolecules, 1999, 32, 290–293 CrossRef CAS.
- C. J. Hawker, D. Mecerreyes, E. Elce, J. Dao, J. L. Hedrick, I. Barakat, P. Dubois, R. Jerome and W. Volksen, “Living free radical polymerization of macromonomers. Preparation of well defined graft copolymers”, Macromol. Chem. Phys., 1997, 198, 155–166 CrossRef CAS.
- I. Capek and M. Akashi, “On the kinetics of free radical polymerization of macromonomers”, J. Macromol. Sci., Part C, 1993, 33, 369–436 CrossRef.
- S. W. Ryu and A. Hirao, “Anionic synthesis of well-defined poly(mhalomethylstyrene)s and branched polymers via graft-onto methodology”, Macromolecules, 2000, 33, 4765–4771 CrossRef CAS.
- M. Gauthier and M. Moeller, “Uniform highly branched polymers by anionic grafting: arborescent graft polymers”, Macromolecules, 1991, 24, 4548–4553 CrossRef CAS.
- M. Schappacher and A. Deffieux, “Newcomblike copolymers of controlled structure and dimensions obtained by grafting polystyryllithium onto poly(chloroethyl vinyl ether) chains”, Macromol. Chem. Phys., 1997, 198, 3953–3961 CrossRef CAS.
- M. Schappacher and A. Deffieux, “New polymer chain architecture: synthesis and characterization of star polymers with comb polystyrene branches”, Macromolecules, 2000, 33, 7371–7377 CrossRef CAS.
- G. H. Fredrickson, “Surfactant-induced lyotropic behavior of flexible polymer solutions”, Macromolecules, 1993, 26, 2825–2831 CrossRef CAS.
- H. Iatrou, J. W. Mays and N. Hadjichristidis, “Regular comb polystyrenes and graft polyisoprene/polystyrene copolymers with double branches (“centipedes”). Quality of (1,3-phenylene)bis(3-methyl-1-phenylpentylidene)dilithium initiator in the presence of polar additives”, Macromolecules, 1998, 31, 6697–6701 CrossRef CAS.
- J. Bernard, M. Schappacher, E. Ammannati, A. Kuhn and A. Deffieux, “Comb and dendrigraft polystyrenes with ferrocenyl units at the periphery: synthesis and electrochemical properties”, Macromolecules, 2002, 35, 8994–9000 CrossRef CAS.
- M. Schappacher, C. Billaud, C. Paulo and A. Deffieux, “Synthesis, dimensions, and solution properties of linear and macrocyclic poly(chloroethyl vinyl ether)-g-polystyrene comblike polymers”, Macromol. Chem. Phys., 1999, 200, 2377–2386 CrossRef CAS.
- D. Lanson, F. Ariura, M. Schappacher, R. Borsali and A. Deffieux, “Application of living ionic polymerizations to the design of AB-type comb-like copolymers of various topologies and organizations”, Macromol. Res., 2007, 15, 173–177 CrossRef CAS.
- M. Gauthier, L. Tichagwa, J. S. Downey and S. Gao, “Arborescent graft copolymers: highly branched macromolecules with a core-shell morphology”, Macromolecules, 1996, 29, 519–527 CrossRef CAS.
- H. Gao and K. Matyjaszewski, “Synthesis of molecular brushes by “grafting onto” method: combination of ATRP and click reactions”, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- B. S. Sumerlin, D. Neugebauer and K. Matyjaszewski, “Initiation efficiency in the synthesis of molecular brushes by grafting from via atom transfer radical polymerization”, Macromolecules, 2005, 38, 702–708 CrossRef CAS.
- R. Venkatesh, L. Yajjou, C. E. Koning and B. Klumperman, “Novel brush copolymers via controlled radical polymerization”, Macromol. Chem. Phys., 2004, 205, 2161–2168 CrossRef CAS.
- S. Muthukrishnan, M. Zhang, M. Burkhardt, M. Drechsler and H. Mori, “Molecular sugar sticks: cylindrical glycopolymer brushes”, Macromolecules, 2005, 38, 7926–7934 CrossRef CAS.
- K. Min, S. Yu, H-i. Lee, L. Mueller, S. S. Sheiko and K. Matyjaszewski, “Yield synthesis of molecular brushes via ATRP in miniemulsion”, Macromolecules, 2007, 40, 6557–6563 CrossRef CAS.
- J. Pietrasik, B. S. Sumerlin, H.-i. Lee, R. R. Gil and K. Matyjaszewski, “Structural mobility of molecular bottle-brushes investigated by NMR relaxation dynamics”, Polymer, 2007, 48, 496–501 CrossRef CAS.
- J. Huang, C. Tang, H. Lee, T. Kowalewski and K. Matyjaszewski, “A novel route for the preparation of discrete nanostructured carbons from block copolymers with polystyrene segments”, Macromol. Chem. Phys., 2007, 208, 2312–2320 CrossRef CAS.
- C. Tang, B. Dufour, T. Kowalewski and K. Matyjaszewski, “Synthesis and morphology of molecular brushes with polyacrylonitrile block copolymer side chains and their conversion into nanostructured carbons”, Macromolecules, 2007, 40, 6199–6205 CrossRef CAS.
- D. Neugebauer, B. S. Sumerlin, K. Matyjaszewski, B. Goodhart and S.S. Sheiko, “How dense are cylindrical brushes grafted from a multifunctional macroinitiator?”, Polymer, 2004, 45, 8173–8179 CrossRef CAS.
- S. Qin, K. Matyjaszewski, H. Xu and S. S. Sheiko, “Synthesis and visualization of densely grafted molecular brushes with crystallizable poly(octadecyl methacrylate) block segments”, Macromolecules, 2003, 36, 605–612 CrossRef CAS.
- S. Rathgeber, T. Pakula, A. Wilk, K. Matyjaszewski and K. L. Beers, “On the shape of bottle-brush macromolecules: systematic variation of architectural parameters”, J. Chem. Phys., 2005, 122, 124904/1-/13 CrossRef.
- S. Rathgeber, T. Pakula, A. Wilk, K. Matyjaszewski, H.-i Lee and K. L. Beers, “Bottle-brush macromolecules in solution: comparison between results obtained from scattering experiments and computer simulations”, Polymer, 2006, 47, 7318–7327 CrossRef CAS.
- S. Rathgeber, H.-i. Lee, K. Matyjaszewski and E. Di Cola, “Rheooscillations of a bottlebrush polymer solution due to shear-induced phase transitions between a shear molten state and a line hexatic phase”, Macromolecules, 2007, 40, 7680–7688 CrossRef CAS.
- O. Prucker and J. Rühe, “Polymer Layers through Self-Assembled Monolayers of Initiators”, Langmuir, 1998, 14, 6893–6898 CrossRef CAS.
- L. Ista, S. Mendez, V. Perez-Luna and G. Lopez, “Synthesis of Poly(N-isopropylacrylamide) on Initiator-Modified Self-Assembled Monolayers”, Langmuir, 2001, 17, 2552–2555 CrossRef CAS.
-
R. Jordan, Surface-Initiated Polymerization I, Springer-Verlag, New York, 2006 Search PubMed.
- G. Sakellariou, M. Park, R. Advincula, J. W. Mays and N. Hadjichristidis, “Homopolymer and Block Copolymer Brushes on Gold by Living Anionic Surface-Initiated Polymerization in a Polar Solvent”, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 769–782 CrossRef CAS.
- R. Advincula, Q. G. Zhou, M. Park, S. G. Wang, J. Mays, G. Sakellariou, S. Pispas and N. Hadjichristidis, “Polymer Brushes by Living Anionic Surface Initiated Polymerization on Flat Silicon (SiOx) and Gold Surfaces: Homopolymers and Block Copolymers”, Langmuir, 2002, 18, 8672–8684 CrossRef CAS.
-
R. Advincula, W. Brittain, K. Caster and J. Rühe, Polymer Brushes: Synthesis, Characterization, Application, Wiley-VCH, Weimheim, Germany, 2004 Search PubMed.
- T. M. Fulghum, N. C. Estillore, C.-D. Vo, S. P. Armes and R. C. Advincula, “Stimuli-Responsive Polymer Ultrathin Films with a Binary Architecture: Combined Layer-by-Layer Polyelectrolyte and Surface-Initiated Polymerization Approach”, Macromolecules, 2008, 41, 429–435 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. Börner, A. Laschewsky, C. Duschl and J.-F. Lutz, “Controlled Cell Adhesion on PEG-Based Switchable Surfaces”, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- J. T. Koberstein, “Molecular design of functional polymer surface”, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 2942–2956 CrossRef CAS.
- F. Liu and M. W. Urban, “Recent advances and challenges in designing stimuli-responsive polymers”, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- F. Xia, Y. Zhu, L. Feng and L. Jiang, “Smart responsive surfaces switching reversibly between super-hydrophobicity and super-hydrophilicity”, Soft Matter, 2009, 5, 275–281 RSC.
- S. Alexander, “Adsorption of chain molecules with a polar head: a scaling prediction”, J. Phys. (Paris), 1997, 38, 983–987 Search PubMed.
- P. Auroy, L. Auvray and L. Leger, “Characterization of the brush regime for grafted polymer layers at the solid-liquid interface”, Phys. Rev. Lett., 1991, 66, 719–722 CrossRef CAS.
- M. Kaholek, W. K. Lee, S. J. Ahn, H. W. Ma, K. C. Caster, B. LaMattina and S. Zauscher, “Stimulus-responsive poly(N-isopropylacrylamide) brushes and nanopatterns prepared by surface-initiated polymerization”, Chem. Mater., 2004, 16, 3688–3696 CrossRef CAS.
- M. K. Vyas, K. Schneider, B. Nandan and M. Stamm, “Switching of friction by binary polymer brushes” Soft Matter, 2008, 4, 1024–1032 CAS.
- B. Zhao, R. T. Haasch and S. MacLaren, “Solvent-induced self-assembly of mixed poly(methyl methacrylate)/polystyrene brushes on planar silica substrates: molecular weight effect”, J. Am. Chem. Soc., 2004, 126, 6124–6134 CrossRef CAS.
- M. Lemieux, D. Usov, S. Minko, M. Stamm, H. Shulha and V. V. Tsukruk, “Reorganization of binary polymer brushes: reversible switching of surface microstructures and nanomechanical properties”, Macromolecules, 2003, 36, 7244–7255 CrossRef CAS.
- Y. Guo and M. G. Moffitt, “Semiconductorquantumdots with environmentally responsive mixed polystyrene/poly(methyl methacrylate) brush layers”, Macromolecules, 2007, 40, 5868–5878 CrossRef CAS.
- J. Draper, I. Luzinov, S. Minko, I. Tokarev and M. Stamm, “Mixed polymerbrushes by sequential polymer addition: Anchoring layer effect,”, Langmuir, 2004, 20, 4064–4075 CrossRef CAS.
- M. Motornov, S. Minko, K. J. Eichhorn, M. Nitschke, F. Simon and M. Stamm, “Reversible tuning of wetting behaviour of polymersurface with responsive polymer brushes”, Langmuir, 2003, 19, 8077–8085 CrossRef CAS.
- S. Santer, A. Kopyshev, J. Donges, H. K. Yang and J. Ruhe, “Dynamically reconfigurable polymer films: Impact on nanomotion”, Adv. Mater., 2006, 18, 2359–2362 CrossRef CAS.
- M. Motornov, R. Sheparovych, I. Tokarev, Y. Roiter and S. Minko, “ Nonwettable thin films from hybrid polymer brushes can be hydrophilic”, Langmuir, 2007, 23, 13–19 CrossRef CAS.
- R. Sheparovych, M. Motornov and S. Minko, “Adapting low-adhesive thin films from mixed polymer brushes”, Langmuir, 2008, 24, 13828–13832 CrossRef CAS.
- M. Müller, “Phase diagram of a mixed polymer brush”, Phys. Rev. E., 2002, 65, 802–805 Search PubMed.
- M. Zhang, T. Breiner, H. Mori and A. H. E. Muller, “Amphiphilic cylindrical brushes with poly(acrylic acid) core and poly(n-butyl acrylate) shell and narrow length distribution”, Polymer, 2003, 44, 1449–1458 CrossRef CAS.
- G. Cheng, A. Boeker, M. Zhang, G. Krausch and A. H. E. Mueller, “Amphiphilic cylindrical core-shell brushes via a “grafting from” process using ATRP”, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- M. O. Gallyamov, B. Tartsch, A. R. Khokhlov, S. S. Sheiko, H. G. Boerner, K. Matyjaszewski and M. Möller, “Conformational dynamics of single molecules visualized in real time by scanning force microscopy: macromolecular mobility on a substrate surface in different vapours”, J. Microsc., 2004, 215, 245–256 CrossRef CAS.
- P. Y. Lai and K. Binder, “Structure and dynamics of polymer brushes near the T point: a Monte-Carlo simulation,”, J. Chem. Phys., 1992, 97, 586–595 CrossRef CAS.
- D. R. M. Williams, “Grafted polymers in bad solvents: octopus surface micelles”, J. Phys. II, 1993, 3, 1313–1318 CrossRef CAS.
- I. Szleifer and M. A. Carignano, “Tethered polymer layers”, Adv. Chem. Phys., 94, 165–260 CrossRef CAS.
- M. O. Gallyamov, B. Tartsch, A. R. Khokhlov, S. S. Sheiko, H. G. Boerner, K. Matyjaszewski and M. Möller, “Real-time scanning force microscopy of macromolecular conformational transitions”, Macromol. Rapid Commun., 2004, 25, 1703–1707 CrossRef CAS.
- M. O. Gallyamov, B. Tartsch B., A. R. Khokhlov, S. S. Sheiko, H. G. Boerner, K. Matyjaszewski and M. Möller, “Reversible collapse of brushlike macromolecules in ethanol and water vapours as revealed by real-time scanning force microscopy”, Chem.–Eur. J., 2004, 10, 4599–4605 CrossRef CAS.
- M. O. Gallyamov, B. Tartsch, P. Mela, H. Börner, K. Matyjaszewski and S. S. Sheiko, “A scanning force microscopy study on the motion of single brush-like macromolecules on a silicon substrate induced by coadsorption of small molecules”, Phys. Chem. Chem. Phys., 2007, 9, 346–352 RSC.
- Y. Qiu and K. Park, “Environment-sensitive hydrogels for drug delivery”, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- D. Schmaljohann, “Thermo- and pH-responsive polymers in drug delivery”, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- S. Dai, P. Ravi and K. C. Tam, “pH-Responsive polymers: synthesis, properties and applications”, Soft Matter, 2008, 4, 435–149 RSC.
- I. Roy and M. N. Gupta, “pH-responsive polymer-assisted refolding of urea- and organic solvent-denatured alpha-chymotrypsin”, Protein Eng., Des. Sel., 2003, 16, 1153–1157 CrossRef CAS.
- D. M. Lynn, M. M. Amiji and R. Langer, “pH-responsive polymer microspheres: rapid release of encapsulated material within the range of intracellular pH”, Angew. Chem., Int. Ed., 2001, 40, 1707–1710 CrossRef CAS.
- N. Murthy, J. Campbell, N. Fausto, A. S. Hoffman and P. S. Stayton, “Design and synthesis of pH-responsive polymeric carriers that target uptake and enhance the intracellular delivery of oligonucleotides”, J. Controlled Release, 2003, 89, 365–374 CrossRef CAS.
- N. Murthy, J. Campbell, N. Fausto, A. S. Hoffman and P. S. Stayton, “Bioinspired pH-responsive polymers for the intracellular delivery of biomolecular drugs”, Bioconjugate Chem., 2003, 14, 412–419 CrossRef CAS.
- C. A. Lackey, O. W. Press, A. S. Hoffman and P. S. Stayton, “A biomimetic pH responsive polymer directs endosomal release and intracellular delivery of an endocytosed antibody complex”, Bioconjugate Chem., 2002, 13, 996–1001 CrossRef CAS.
- F. A. Plamper, H. Becker, M. Lanzendoerfer, M. Patel and A. Wittemann, “Synthesis, characterization and behavior in aqueous solution of star-shaped poly(acrylic acid)”, Macromol. Chem. Phys., 2005, 206, 1813–1825 CrossRef CAS.
- A. Laguecir, S. Ulrich, J. Labille, N. Fatin-Rouge and S. Stoll, “Size and pH effect on electrical and conformational behavior of poly(acrylic acid): simulation and experiment”, Eur. Polym. J., 2006, 42, 1135–1144 CrossRef CAS.
- H. Lee, J. R. Boyce, A. Nese, S. S. Sheiko and K. Matyjaszewski, “pH-induced conformational changes of loosely grafted molecular brushes containing poly(acrylic acid) side chains”, Polymer, 2008, 49, 5490–5496 CrossRef CAS.
- Y. Xu, S. Bolisetty, M. Drechsler, B. Fang, J. Yuan, M. Ballauff and A. H. E. Müller, “pH and salt responsive poly(N,N-dimethylaminoethyl methacrylate) cylindrical brushes and their quaternized derivatives”, Polymer, 2008, 49, 3957–3964 CrossRef CAS.
- M. Wang, S. Zou, G. Guerin, L. Shen and K. A. Deng, “Water-soluble pH responsive molecular brush of poly(N,N-dimethylaminoethyl methacrylate) grafted polythiophene”, Macromolecules, 2008, 41, 6993–7002 CrossRef CAS.
- E. S. Gil and S. M. Hudson, “Stimuli-reponsive polymers and their bioconjugates”, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, “Bioconjugates of smart polymers and proteins: synthesis and applications”, Macromol. Symp., 2004, 207, 139–151 CrossRef CAS.
- J.-F. Lutz, “Solution Self-Assembly of Tailor-made Macromolecular Building-Blocks Prepared by Controlled Radical Polymerization Techniques”, Polym. Int., 2006, 55, 979–993 CrossRef CAS.
- M. Yamato, O. H. Kwon, M. Hirose, A. Kikuchi, T. Okano and J. Biomed, “Novel patterned cell coculture utilizing thermally responsive grafted polymer surfaces”, Mater. Res., 2000, 55, 137–140 Search PubMed.
- N. Nath and A. Chilkoti, “Fabrication of a reversible protein array directly from cell lysate using a stimuli-responsive polypeptide”, Anal. Chem., 2003, 75, 709–715 CrossRef CAS.
- X. Cheng, Y. Wang, Y. Hanein, K. F. Bohringer and B. D. Ratner, “Novel cell patterning using microheater-controlled thermoresponsive plasma films”, J. Biomed. Mater. Res., 2004, 70A, 159–168 CrossRef CAS.
- X.-Z. Zhang, P. J. Lewis and C.-C. Chu, “Fabrication and characterization of a smart drug delivery system: microsphere in hydrogel”, Biomaterials, 2005, 26, 3299–3309 CrossRef CAS.
- Y. H. Bae, T. Okano, R. Hsu and S. W. Kim, “Thermosensitive polymers as on-off switches for drug release”, Makromol. Chem. Rapid Commun., 1987, 8, 481–485 CrossRef CAS.
- J. Shi, L. Liu, X. Liu, X. Sun and S. Cao, “Inorganic–organic hybrid alginate beads with LCST near human body temperature for sustained dualsensitivedrug delivery”, Polym. Adv. Technol., 2008, 19, 1467–1473 CAS.
- M. N. Albarghouthi and A. E. Barron, “Polymeric matrices for DNA sequencing by capillary electrophoresis”, Electrophoresis, 2000, 21, 4096–4111 CrossRef CAS.
- B. A. Buchholz, E. A. Doherty, M. N. Albarghouthi, F. M. Bogdan and J. M. Zahn, “Microchannel DNA sequencing matrices with a thermally controlled ‘viscosity switch',”, Anal. Chem., 2001, 73, 157–164 CrossRef CAS.
- B. A. Buchholz, W. Shi and A. E. Barron, “Microchannel DNA sequencing matrices with switchable viscosities”, Electrophoresis, 2002, 23, 1398–1409 CrossRef CAS.
- J.-F. Lutz, “Thermo-Switchable Materials Prepared Using the OEGMA-Platform”, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- H. G. Schild, “Poly(N-isopropylacrylamide): experiment, theory and application”, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- X. Wang, X. Qiu and C. Wu, “Comparison of the coil-to-globule and the globule-to-coil transitions of a single poly(N-isopropylacrylamide) homopolymer chain in water”, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- R. Pelton, “Temperature-sensitive aqueous microgels”, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS.
- O. Azzaron, A. A. Brown and W. T. S. Huck, “UCST wetting transitions of polyzwitterionic brushes driven by self-association”, Angew. Chem., Int. Ed., 2006, 45, 1770–1774 CrossRef.
- N. Cheng, A. A. Brown, O. Azzaroni and W. T. S. Huck, “Thickness-dependent properties of polyzwitterionic brushes”, Macromolecules, 2008, 41, 6317–6321 CrossRef CAS.
- T. L. Sun, G. J. Wang, L. Feng, B. Q. Liu, Y. M. Ma, L. Jiang and D. Zhu, “Reversible switching between superhydrophilicity and superhydrophobicity”, Angew. Chem., Int. Ed., 2004, 43, 357–360 CrossRef CAS.
- Q. Fu, G. V. R. Rao, S. B. Basame, D. J. Keller, K. Artyushkova, J. E. López and G. P. Fulghum, “Reversible control of free energy and topography of nanostructured surfaces”, J. Am. Chem. Soc., 2004, 126, 8904–8905 CrossRef CAS.
- T. L. Sun, L. Feng, X. F. Gao and L. Jiang, “Bioinspired surfaces with special wettability”, Acc. Chem. Res., 2005, 38, 644–652 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, “Point by point comparison of two thermosensitive polymers exhibiting a similar LCST: is the age of poly(NIPAM) over?”, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- J.-F. Lutz and A. Hoth, “Preparation of ideal PEG analogues with a tunable thermosensitivity by controlled radical copolymerization of 2-(2-methoxyethoxy)ethyl methacrylate and oligo(ethylene glycol) methacrylate”, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- J.-F. Lutz, “Polymerization of oligo(ethylene glycol) (meth)acrylates: toward new generations of smart biocompatible materials”, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. A. Jones, N. Novo, K. Flagler, C. D. Pagnucco, S. Carew, C. Cheong, X. Z. Kong, N. A. D. Burke and H. D. H. Stöover, “Thermoresponsive Copolymers of Methacrylic Acid and Poly(ethylene glycol) Methyl Ether Methacrylate”, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6095–6104 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, O. Akdemir and A. Hoth, “About the phase transitions in aqueous solutions of thermoresponsive copolymers and hydrogels based on 2-(2-methoxyethoxy)ethyl methacrylate and oligo(ethylene glycol) methacrylate”, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- A. Munoz-Bonilla, M. Fernandez-Garcia and D. M. Haddleton, “Synthesis and aqueous solution properties of stimuli-responsive triblock copolymers”, Soft Matter, 2007, 3, 725–731 RSC.
- K. Skrabania, J. Kristen, A. Laschewsky, O. Akdemir, A. Hoth and J.-F. Lutz, “Design, synthesis, and aqueous aggregation behavior of nonionic single and multiple thermoresponsive polymers”, Langmuir, 2007, 23, 84–93 CrossRef CAS.
- S.-i. Yamamoto, J. Pietrasik and K. Matyjaszewski, “ATRP synthesis of thermally responsive molecular brushes from oligo(ethylene oxide) methacrylates”, Macromolecules, 2007, 40, 9348–9353 CrossRef CAS.
- F. Liu and M. W. Urban, “New thermal transitions in stimuli-responsive copolymer films”, Macromolecules, 2009, 42, 2161–2167 CrossRef CAS.
-
(a) J. E. Comrie and W. T. S. Huck, “Exploring actuation and mechanotransduction properties of polymer brushes”, Macromol. Rapid Commun., 2008, 29, 539–546 CrossRef CAS;
(b) J. E. Comrie and W. T. S. Huck, “Formation of hybrid 2D polymer–metal microobjects,”, Langmuir, 2007, 23, 1569–1576 CrossRef CAS.
- F. Zhou, W. M. Shu, M. E. Welland and W. T. S. Huck, “Highly reversible and multi-stage cantilever actuation driven by polyelectrolyte brushes”, J. Am. Chem. Soc., 2006, 128, 5326–5327 CrossRef CAS.
- A. Valiaev, N. I. Abu-Lail, D. W. Lim, A. Chilkoti and S. Zauscher, “Microcantilever sensing and actuation with end-grafted stimulusresponsive elastin-like polypeptides”, Langmuir, 2007, 23, 339–344 CrossRef CAS.
-
B. Bhushan, Micro/Nanotribology and its applications, Kluwer Academic Press, Dordrecht, The Netherlands, 1997 Search PubMed.
-
B. Bhushan, Tribology issues and opportunities in MEMS, Kluwer Academic Press, Dordrecht, The Netherlands, 1997 Search PubMed.
- S. Santer, A. Kopyshev, J. Donges, H.-K. Yang and J. Rühe, “Domain Memory of Mixed Polymer Brushes”, Langmuir, 2006, 22, 4660–4667 CrossRef CAS.
- V. V. Tsukruk, “Molecular Lubricants and Glues for Micro- and Nanodevices”, Adv. Mater., 2001, 13, 95–108 CrossRef CAS.
- L. Ionov, S. Minko, M. Stamm, J. F. Gohy, R. Jérôme and A. Scholl, “Reversible Chemical Patterning on Stimuli Responsive Polymer Film - Environment Responsive Lithography”, J. Am. Chem. Soc., 2003, 125, 8302–8306 CrossRef CAS.
- Y. Liu, L. Mu, B. H. Liu and J. L. Kong, “Controlled Switchable Surface”, Chem.–Eur. J., 2005, 11, 2622–2631 CrossRef CAS.
- D. M. Jones, R. R. Smith, W. T. S. Huck and C. Alexander, “Variable adhesion of micropatterned thermoresponsive polymer brushes: AFM investigations of poly (N-isopropylacrylamide) brushes prepared by surface-initiated polymerizations”, Adv. Mater., 2002, 14, 1130–1134 CrossRef CAS.
- I. A. Aksay, M. Trau, S. Manne, I. Honma, N. Yao, L. Zhou, P. Fenter, P. M. Eisenberger and S. M. Gruner, “Biomimetic Pathways for Assembling Inorganic Thin Films”, Science, 1996, 273, 892–898 CAS.
- S. S. Sheiko, S. A. Prokhorova, K. L. Beers, K. Matyjaszewski, I. I. Potemkin, A. R. Khokhlov and M. Möller, “Single molecule rod-globule phase transition for brush molecules at a flat interface,”, Macromolecules, 2001, 34, 8354–8360 CrossRef CAS.
- F. Sun, S. S. Sheiko, M. Moeller, K. Beers and K. Matyjaszewski, “Conformational switching of molecular brushes in response to the energy of interaction with the substrate”, J. Phys. Chem. A, 2004, 108, 9682–9686 CrossRef CAS.
- I. I. Potemkin, A. R. Khokhlov, S. Prokhorova, S. S. Sheiko, M. Moeller and K. L. Beers, “Spontaneous curvature of comblike polymers at a flat interface”, Macromolecules, 2004, 37, 3918–3923 CrossRef CAS.
- H. Xu, F. C. Sun, D. G. Shirvanyants, M. Rubinstein, D. Shabratov, K. L. Beers, K. Matyjaszewski and S. S. Sheiko, “Molecular pressure sensors”, Adv. Mater., 2007, 19, 2930–2934 CrossRef CAS.
- S. V. Panyukov, S. S. Sheiko and M. Rubinstein, “Amplification of tension in branched macromolecules,”, Phys. Rev. Lett., 2009, 102, 148301/1-/4 CrossRef.
- S. Panyukov, E. B. Zhulina, S. S. Sheiko, G. C. Randall and J. Brock, “Tension amplification in molecular brushes in solutions and on substrates”, J. Phys. Chem. B, 2009, 113, 3750–3768 CrossRef CAS.
- N. V. Lebedeva, F. C. Sun, H.-I. Lee, K. Matyjaszewski and S. S. Sheiko, “Fatal adsorption” of brushlike macromolecules: high sensitivity of C–C bond cleavage rates to substrate surface energy”, J. Am. Chem. Soc., 2008, 130, 4228–4229 CrossRef CAS.
- I. Park, S. S. Sheiko, A. Nese and K. Matyjaszewski, “Molecular tensile testing machines: breaking a specific covalent bond by adsorption induced tension in brush like macromolecules”, Macromolecules, 2009, 42, 1805–1807 CrossRef CAS.
- A. K. Anal, “Stimuli-induced pulsatile or triggered release delivery systems for bioactive compounds”, Recent Pat. Endocr., Metab. Immune Drug Discovery, 2007, 1, 83–90 CrossRef CAS.
- T. Shiga, “Deformation and viscoelastic behavior of polymer gels in electric fields”, Adv. Polym. Sci., 1997, 134, 31–63 CrossRef.
- R. Kroeger, H. Menzel and M. L. Hallensleben, “Light controlled solubility change of polymers: Copolymers of N,N-dimethylacrylamide and 4-phenylazophenyl acrylate”, Macromol. Chem. Phys., 1994, 195, 2291–2298 CrossRef CAS.
- H. Akiyama and N. Tamaoki, “Polymers derived from N-isopropylacrylamide and azobenzene-containing acrylamides: Photoresponsive affinity to water,”, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5200–5214 CrossRef CAS.
- D. Kungwatchakun and M. Irie, “Photoresponsive polymers. Photocontrol of the phase separation temperature of aqueous solutions of poly-[N-isopropylacrylamide-co-N-(4-phenylazophenyl)acrylamide]”, Macromol. Chem., Rapid. Commun., 1998, 9, 243–246 Search PubMed.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, “Photoresponsive polymer–enzyme switches”, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS.
- F. D. Jochum and P. Theato, “Temperature and Light Sensitive Copolymers Containing Azobenzene Moieties Prepared via a Polymer Analogous Reaction Polymer”, Polymer, 2009, 50, 3079–3085 CrossRef CAS.
- K. G. Yager and C. J. Barrett, “Novel photo-switching using azobenzene functional materials,”, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- C. Luo, F. Zuo, X. Ding, Z. Zheng, X. Cheng and Y. J. Peng, “Light-triggered reversible solubility of α-cyclodextrin and azobenzene moiety complexes in PDMAA-co-PAPA via molecular recognition”, J. Appl. Polym. Sci., 2008, 107, 2118–2125 CrossRef CAS.
- H. Akiyama and N. Tamaoki, “Synthesis and Photoinduced Phase Transitions of Poly(N-isopropylacrylamide) Derivative Functionalized with Terminal Azobenzene Units”, Macromolecules, 2007, 40, 5129–5132 CrossRef CAS.
- I. Lokuge, X. Wang and P. W. Bohn, “Temperature-controlled flow switching in nanocapillary array membranes mediated by poly(Nisopropylacrylamide) polymer brushes grafted by atom transfer radical polymerization”, Langmuir, 2007, 23, 305–311 CrossRef CAS.
- C. S. Kwok, P. D. Mourad, L. A. Crum and B. D. Ratner, “Self-assembled molecular structures as ultrasonically-responsive barrier membranes for pulsatile drug delivery”, J. Biomed. Mater. Res., 2001, 57, 151–164 CrossRef CAS.
- S. Tugulu, M. Harms, M. Fricke, D. Volkmer and H.A. Klok, “Polymer brushes as ionotropic matrices for the directed fabrication of microstructured calcite thin films”, Angew. Chem., Int. Ed., 2006, 45, 7458–6741 CrossRef CAS.
- R. Tadmor, J. Janik, J. Klein and L. J. Fetters, “Sliding Friction with Polymer Brushes”, Phys. Rev. Lett., 2003, 91, 115503–1-4 CrossRef.
- T. Moro, Y. Takatori, K. Ishihara, T. Konno, Y. Takigawa, T. Matsushita, U. Chung, K. Nakamura and H. Kawaguchi, “Surface grafting of artificial joints with a biocompatible polymer for preventing periprosthetic osteolysis”, Nat. Mater., 2004, 3, 829–836 CrossRef CAS.
- J. Klein, “Repair or Replacement-A Joint Perspective”, Science, 2009, 323, 47–48 CrossRef CAS.
- A. Galuschko, L. Spirin, T. Kreer, A. Johner, C. Pastorino, J. Wittmer and J. Baschnagel, “ Frictional Forces between Strongly Compressed, Nonentangled Polymer Brushes: Molecular Dynamics Simulations and Scaling Theory”, Langmuir, 2010, 26, 6418–6429 CrossRef CAS.
- L. Spirin, A. Galuschko, T. Kreer, A. Johner, J. Baschnagel and K. Binder, “Polymer-brush lubrication in the limit of strong compression”, Eur. Phys. J. E, 2010, 33, 307–311 CrossRef CAS.
- L. Spirin, A. Galuschko, T. Kreer, K. Binder and J. Baschnagel, “Polymer-Brush Lubricated Surfaces with Colloidal Inclusions under Shear Inversion”, Phys. Rev. Lett., 2011, 106, 168301–1-4 CrossRef CAS.
- J. Klein, “Molecular mechanisms of synovial joint lubrication”, Proc Inst. Mech. Eng., Part J, 2006, 220, 691–710 CAS.
- N. Nath and A. Chilkoti, “Creating ‘smart’ surfaces using stimuli responsive polymers”, Adv. Mater., 2002, 14, 1243–1247 CrossRef CAS.
- J. H. Zhou, G. Wang, J. Q. Hu, X. B. Lu and J. H. Li, “Temperature, ionic strength and pH induced electrochemical switching of smart polymer interfaces”, Chem. Commun., 2006, 4820–4822 RSC.
- Y. D. Mao, C. X. Luo and Q. Ouyang, “Studies of temperature-dependent electronic transduction on DNA hairpin loop sensor”, Nucleic Acids Res., 2003, 31, e108 CrossRef.
- F. Ricci, R.Y. Lai, A. J. Heeger, K. W. Plaxco and J. J. Sumner, “Effect of Molecular Crowding on the Response of an Electrochemical DNA Sensor”, Langmuir, 2007, 23, 6827–6834 CrossRef CAS.
- C. E. Immoos, S. J. Lee and M. W. Grinstaff, “Conformationally Gated Electrochemical Gene Detection”, ChemBioChem, 2004, 5, 1100–1103 CrossRef CAS.
- S. Tyagi and F. R. Kramer, “Molecular Beacons: Probes that Fluoresce upon Hybridization”, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- C. Fan, K. W. Plaxco and A. J. Heeger, “Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA”, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9134–9137 CrossRef CAS.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, “Label-Free Electronic Detection of Thrombin in Blood Serum by Using an Aptamer-Based Sensor”, Angew. Chem., Int. Ed., 2005, 44, 5456–5459 CrossRef CAS.
- A. E. Radi, J. L. A. Sanchez, E. Baldrich and C. K. O'Sullivan, “Reagentless, Reusable, Ultrasensitive Electrochemical Molecular Beacon Aptasensor”, J. Am. Chem. Soc., 2006, 128, 117–124 CrossRef CAS.
- T. Hermann and D. J. Patel, “Adaptive Recognition by Nucleic Acid Aptamers”, Science, 2000, 287, 820–825 CrossRef CAS.
- B. R. Baker, R. Y. Lai, M. S. Wood, E. H. Doctor, A. J. Heeger and K. W. Plaxco, “An Electronic, Aptamer-Based Small-Molecule Sensor for the Rapid, Label-Free Detection of Cocaine in Adulterated Samples and Biological Fluids”, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS.
- X. L. Zuo, S. P. Song, J. Zhang, D. Pan, L. H. Wang and C. Fan, “A Target-Responsive Electrochemical Aptamer Switch (TREAS) for Reagentless Detection of Nanomolar ATP”, J. Am. Chem. Soc., 2007, 129, 1042–1043 CrossRef CAS.
- J. H. Collier and M. Mrksich, “Engineering a biospecific communication pathway between cells and electrodes”, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2021–2025 CrossRef CAS.
- W. S. Yeo and M. Mrksich, “Self-Assembled Monolayers That Transduce Enzymatic Activities to Electrical Signals”, Angew. Chem., Int. Ed., 2003, 42, 3121–3124 CrossRef CAS.
- M. Zrinyi, “Intelligent polymer gels controlled by magnetic fields”, Colloid Polym. Sci., 2000, 278, 98–103 CAS.
- K. D. Harris, R. Cuypers, P. Scheibe, C. L. van Oosten, C. W. M. Bastiaansen, J. Lub and D. J. Broer, “Large amplitude light-induced motion in high elastic modulus polymer actuators”, J. Mater. Chem., 2005, 15, 5043–5048 RSC.
- S. Juodkazis, N. Mukai, R. Wakaki, A. Yamaguchi, S. Matsuo and H. Misawa, “Reversible phase transitions in polymer gels induced by radiation forces”, Nature, 2000, 408, 178–181 CrossRef CAS.
- A. Kikuchi and T. Okano, “Pulsatile drug release control using hydrogels”, Adv. Drug Delivery Rev., 2002, 54, 53–77 CrossRef CAS.
- M. Nakayama, T. Okano, T. Miyazaki, F. Kohori, K. Sakai and M. Yokoyama, “Molecular design of biodegradable polymeric micelles for temperature-responsive drug release”, J. Controlled Release, 2006, 115, 46–56 CrossRef CAS.
- L. E. Bromberg and E. S. Ron, “Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery”, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS.
- L. D. Taylor and L. D. Cerankowski, “Preparation of films exhibiting a balanced temperature dependence to permeation by aqueous solutions: a study of lower consolute behavior”, J. Polym. Sci. A, 1975, 13, 2551–2570 CAS.
- S. Fujishige, K. Kubota and L. Ando, “Phase transition of aqueous solutions of poly(N-isopropylacrylamide) and poly(N-isopropylmethacrylamide)”, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- K. Kubota, S. Fujishige and I. Ando, “Single-chain transition of poly(N-isopropylacrylamide) in water”, J. Phys. Chem., 1990, 94, 5154–5158 CrossRef CAS.
- H. Inomata, S. Goto and S. Saito, “Phase transition of N-substituted acrylamide gels”, Macromolecules, 1990, 23, 4887–4888 CrossRef CAS.
- M. M. Prange, H. H. Hooper and J. M. Prausnitz, “Thermodynamics of aqueous systems containing hydrophilic polymers or gels”, AIChE J., 1989, 35, 803–813 CrossRef CAS.
- N. T. Southall, K. A. Dill and A. D. J. Haymet, “A view of the hydrophobic effect”, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS.
- W. S. Yeo, M. N. Yousaf and M. Mrksich, “ Dynamic Interfaces between Cells and Surfaces: Electroactive Substrates that Sequentially Release and Attach Cells”, J. Am. Chem. Soc., 2003, 125, 14994–14995 CrossRef CAS.
- M. Gerard, A. Chaubey and B. D. Malhotra, “Application of conducting polymers to biosensors”, Biosens. Bioelectron., 2002, 17, 345–359 CrossRef CAS.
- P. M. George, D. A. LaVan, J. A. Burdick, C. Y. Chen, E. Liang and R. Langer, “Electrically Controlled Drug Delivery from Biotin-Doped Conductive Polypyrrole”, Adv. Mater., 2006, 18, 577–581 CrossRef CAS.
- B. Zinger and L. L. Miller, “Timed release of chemicals from polypyrrole films”, J. Am. Chem. Soc., 1984, 106, 6861–6863 CrossRef CAS.
- J. Y. Wong, R. Langer and D. E. Ingber, “Electrically conducting polymers can noninvasively control the shape and growth of mammalian cells”, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3201–3204 CrossRef CAS.
- Y. Liu, L. Mu, B. H. Liu, S. Zhang, P. Y. Yang and J. L. Kong, “Controlled protein assembly on a switchable surface”, Chem. Commun., 2004, 1194–1195 RSC.
- L. Mu, Y. Liu, S. Y. Cai and J. L. Kong, “A Smart Surface in a Microfluidic Chip for Controlled Protein Separation”, Chem.–Eur. J., 2007, 13, 5113–5120 CrossRef CAS.
- I. Tokarev and S. Minko, “Multiresponsive hierarchically structured membranes: new challenging biomimetic materials for biosensors, controlled release, biochemical gates and nanoreactors”, Adv. Mater., 2009, 21, 241–247 CrossRef CAS.
- I. Tokarev, V. Gopishetty, J. Zhou, M. Pita, M. Motornov, E. Katz and S. Minko, Stimuli-responsive hydrogel membranes coupled with biocatalytic processes”, ACS Appl. Mater. Interfaces, 2009, 1, 532–536 CAS.
- M. Motornov, T. K. Tam, M. Pita, I. Tokarev, E. Katz and S. Minko, “Switchable selectivity for gating ion transport with mixed polyelectrolyte brushes: approaching ‘smart’ drug delivery systems”, Nanotechnology, 2009, 20, 434006–434016 CrossRef.
- V. N. Wong, G. Fernando, A. R. Wagner, J. Zhang, G. R. Kinsel, S. Zauscher and D. J. Dyer, “Separation of peptides with polyionic nanosponges for MALDI-MS analysis”, Langmuir, 2009, 25, 1459–1465 CrossRef CAS.
- S. J. Lue, J. J. Hsu and T. C. Wei, “Drug permeation modeling through the thermo-sensitive membranes of poly(N-isopropylacrylamide) brushes grafted onto micro-porous films”, J. Membr. Sci., 2008, 321, 146–154 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, “Effects of graft densities and chain lengths on separation of bioactive compounds by nanolayered thermoresponsive polymer brush surfaces”, Langmuir, 2008, 24, 511–517 CrossRef CAS.
- Q. Yang, J. Tian, M. X. Hu and Z. K. Xu, “Construction of a comb-like glycosylated membrane surface by a combination of UV induced graft polymerization and surface-initiated ATRP”, Langmuir, 2007, 23, 6684–6690 CrossRef CAS.
- B. Yang and W. T. Yang, “Thermo-sensitive switching membranes regulated by pore-covering polymer brushes”, J. Membr. Sci., 2003, 218, 247–255 CrossRef CAS.
- H. J. Zhang and Y. Ito, “pHcontrol of transport through a porous membrane self-assembled with a poly(acrylic acid) loop brush”, Langmuir, 2001, 17, 8336–8340 CrossRef CAS.
- M. Ulbricht and H. Yang, “Controlled Switchable Surface”, Chem. Mater., 2005, 17, 2622–2631 CrossRef CAS.
- L. Ying, E. T. Kang, K. G. Neoh, K. Kato and H. Iwata, “Drug permeation through temperature-sensitive membranes prepared from poly(vinylidene fluoride) with grafted poly(N-isopropylacrylamide) chains”, J. Membr. Sci., 2004, 243, 253–262 CrossRef CAS.
- A. M. Granville and W. J. Brittain, “Stimuli-responsive semi-fluorinated polymer brushes on porous silica substrates”, Macromol. Rapid Commun., 2004, 25, 1298–1302 CrossRef CAS.
- A. Iwanade, D. Umeno, K. Saito and T. Sugo, “Protein binding to amphoteric polymer brushes grafted onto a porous hollow-fiber membrane”, Biotechnol. Prog., 2007, 23, 1425–1430 CrossRef CAS.
- P. Jain, L. Sun, J. H. Dai, G. L. Baker and M. L. Bruening, “High-capacity purification of his-tagged proteins by affinity membranes containing functionalized polymer brushes”, Biomacromolecules, 2007, 8, 3102–3107 CrossRef CAS.
- Y. Lu, Y. Mei and M. Ballauff, “Thermosensitive Core-Shell Particles as Carrier Systems for Metallic Nanoparticles”, J. Phys. Chem. B, 2006, 110, 3930–3937 CrossRef CAS.
- N. Estillore and R. Advincula, “Solvent-Responsive and Free-Standing Films of Semi-Fluorinated Block Copolymer Brushes via Layer-by-Layer Deposited Polyelectrolyte Macroinitiators”, Macromol. Chem. Phys., 2011, 212, 1552–1566 CrossRef CAS.
- N. Yamada, T. Okano, H. Sakai, F. Karikusa, T. Sawasaki and Y. Sakurai, “Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells”, Makromol. Chem. Rapid Commun., 1990, 11, 571–576 CrossRef CAS.
- H. J. Lee, S. E. Kim, I. K. Kwon, C. Park, C. Kim, J. Yang and S. C. Lee, “Spatially mineralized self-assembled polymeric nanocarriers with enhanced robustness and controlled drug-releasing property”, Chem. Commun., 2010, 46, 345–492 RSC.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.