Optically controlled liquid flow in initially prohibited elastomeric nanocomposite micro-paths†
Francesca
Villafiorita-Monteleone
*ab,
Elisa
Mele
*a,
Gianvito
Caputo
b,
Fabrizio
Spano
a,
Salvatore
Girardo
b,
P. Davide
Cozzoli
bc,
Dario
Pisignano
*bc,
Roberto
Cingolani
d,
Despina
Fragouli
a and
Athanassia
Athanassiou
*abd
aCenter for Biomolecular Nanotechnologies, Istituto Italiano di Tecnologia @UniLe, via Barsanti, 73010 Arnesano, Lecce, Italy. E-mail: francesca.villafiorita@unisalento.it; elisa.mele@iit.it; fabrizio.spano@iit.it; despina.fragouli@iit.it; athanassia.athanassoiu@iit.it
bNational Nanotechnology Laboratory, Istituto Nanoscienze-CNR, via per Arnesano, 73100 Lecce, Italy. E-mail: gianvito.caputo@unisalento.it; salvatore.girardo@unisalento.it; davide.cozzoli@unisalento.it; dario.pisignano@unisalento.it
cDipartimento di Matematica e Fisica “Ennio De Giorgi”-Università del Salento, via per Arnesano, 73100 Lecce, Italy
dIstituto Italiano di Tecnologia (IIT), via Morego 30, 16163 Genova, Italy. E-mail: roberto.cingolani@iit.it
First published on 7th August 2012
Abstract
The significant increment of TiO2 surface wettability upon UV irradiation makes it a promising component of materials or systems with tunable surface wetting characteristics. This remarkable property of TiO2 is retained in the nanocomposite materials developed for this work, which consist of the elastomer PDMS enriched with organic-capped nanorods of TiO2. In particular, the nanocomposites demonstrate a surface transition from a hydrophobic state to a hydrophilic one under selective pulsed UV laser irradiation. This wettability change is reversible, with the hydrophobic character of the nanocomposites being fully recovered after a couple of days of samples storage in moderate vacuum. The hydrophobic-to-hydrophilic transition and recovery can be repeated tens of times on the same sample without any apparent fatigue. As verified by XPS and AFM analysis, the wettability enhancement is exclusively attributed to the TiO2 nanorods exposed on the nanocomposite surface. The tuning of the surface wettability properties of the PDMS/TiO2 materials, together with the easy processability of this elastomer, opens the way to the realization of microfluidic devices with controlled liquid flow. We demonstrate the potentiality of such systems by fabricating microfluidic channels with walls of PDMS and PDMS/TiO2 nanorods composite materials. The combination of the used geometry with the hydrophobic character of both the pure and nanocomposite PDMS prohibits the penetration of water in their developed microchannels. After UV irradiation, water penetration is allowed inside the irradiated nanocomposite microfluidic channels, whereas it is still forbidden after the irradiation of the bare PDMS microchannels, revealing the essential role of the TiO2 nanofillers.
Introduction
In recent times, research efforts have been focused on the design and manufacturing of “smart” materials characterized by the reversible change of their surface properties upon the application of an external stimulus (pH, temperature, voltage, light, etc.).1,2 One of the most frequently adopted solutions is the use of photoresponsive components for the development of surfaces with reversible wettability. These components can be classified in two main groups. The first one is the group of photochromic molecules, which switch reversibly between two stable isomers when irradiated with light of specific wavelengths. The alteration of the wetting properties of photochromic monolayers or microcrystalline films can be due to changes in the orientation of the molecules or in the size distribution of the microcrystals, respectively, as demonstrated with very small (≈4°) or quite large (≈35°) variations of the water contact angle (WCA).3,4 When photochromic molecules are incorporated into polymeric matrices, by functionalization or simple mixing, in order to develop solid materials, the typical variation in WCA is due to polarity changes in the photochromic isomers, and is usually around 10°, a value that can be slightly extended by enhancing the roughness of the composite surface.5–7 The second group of photoresponsive components is one of inorganic semi-conductor oxides such as TiO2, ZnO, WO3, V2O5, SnO2. On surfaces, upon bandgap photoexcitation, oxygen vacancies are created, promoting the dissociative adsorption of water, and consequently increasing their hydrophilicity.8,9 The wettability changes in materials based on such oxides vary considerably from very small (<10°) to extremely large (≈180°), depending on the examined system and on the involved superficial topography.10–13Both photochromic molecules and inorganic oxides have been exploited for the development of a special class of photoresponsive materials, on the surface of which directional liquid drop movement has been observed, due to a light induced surface energy gradient.14–16 Our group recently demonstrated the successful incorporation of colloidal titanium dioxide (TiO2) nanorods (NRs) into a poly(methyl methacrylate) matrix for the realization of surface wettability gradients suitable for the spontaneous movement of water droplets.16 The optically driven liquid drop motion represents an alternative strategy to those already adopted for transporting and manipulating small liquid volumes (from pL to μL) also within microchannels, such as mechanical or optical pumps and valves,17,18 or thermal and chemical methods.19,20 Moreover, it is very appealing in microfluidic devices for biomedical, chemical or optical applications, which target miniaturization and automation of operating elements, to have flexibility in design, and low costs.21 Towards this approach, Caprioli et al.22 demonstrated the successful realization of microchannels made of a cyclic olefin copolymer (TOPAS) doped with a photochromic compound. The wettability changes of the capillary walls obtained upon irradiation with ultraviolet (UV) light have been employed to control the penetration dynamics of hydrophilic polyurethane.
Targeting microfluidic applications with photoinduced spontaneous liquid flow, we present herein a composite system based on NRs of the most studied photoresponsive inorganic oxide, TiO2, and the elastomer of choice in microfluidics, poly(dimethylsiloxane) (PDMS). PDMS shows interesting properties, like low elastic modulus, non-toxicity, high permeability to gases, physical and chemical stability over a wide range of temperatures (−50 °C to 200 °C),23 and easy processability.24 On the other hand, its highly hydrophobic character (WCA around 110°) prohibits the spontaneous filling of aqueous solutions into microchannels.25 To enhance its wettability, different techniques have been adopted so far, based on the modification of the material surface in a reversible or irreversible way.26,27 The combination of PDMS with light sensitive TiO2 is so far exploited by only a few groups. Nakata et al.24 prepared amorphous TiO2–PDMS composite films by a sol–gel method followed by curing with UV light. In this case, boiling water treatment was essential for the crystallization of TiO2 in order to exhibit the desired photoinduced superhydrophilicity under UV irradiation. The strategy cannot be directly applied to microfluidics, since hot water had to be previously forced in the capillaries to form the crystalline TiO2–PDMS composite walls. Nagai et al.26 described a microfluidic device consisting of a structured PDMS mold placed in contact with a TiO2-coated quartz substrate, which is based on centrifugal forces to drive the liquid penetration into the channels. The photocatalytic properties of TiO2were used to release hydrophobic material from the polymer after irradiation with UV lamp, with a consequent increment in WCA of the TiO2 surface.
The approach presented herein is focused on the spontaneous penetration of water in initially forbidden PDMS nanocomposite microfluidic paths. Specifically, we describe the use of chemically synthesized colloidal anatase TiO2 NRs embedded in a PDMS matrix to produce microfluidic devices realized by soft lithographic procedures with optically-controlled wettability changes. Water penetration is completely forbidden in the developed PDMS and PDMS/TiO2 microchannels, due to the hydrophobic nature of the involved surfaces. Water penetrates spontaneously only in the PDMS/TiO2 microchannels after pulsed laser UV irradiation, since the walls of the microchannels become hydrophilic. By using spectroscopic analysis and microscope imaging, we prove that the TiO2 NRs exposed on the surface of our composite material are responsible for the UV-induced hydrophilicity. The presented materials and systems offer the opportunity to fully control liquid penetration flow in fluidic devices, without the use of external mechanical parts, useful in applications for bioanalysis,28 examination and manipulation of single cells or molecules,29 multiphase flows,30 and drug delivery.31
Experimental
Materials
All chemicals were used as received. PDMS (Sylgard 184) was purchased by Dow Corning, Midland, MI. Toluene solutions of oleic acid-capped TiO2 NRs with a mean length of 20 nm and an average diameter of 3 nm were prepared as described elsewhere.32 24 × 24 mm2 glass microscope slides (Forlab, Carlo Erba) were used to create the fourth wall in the microfluidic system. All solvents used were purchased from Carlo Erba Reagenti.Preparation of the nanocomposite microfluidic systems
Solutions of PDMS (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base:curing agent) and TiO2 NRs in toluene were prepared at concentrations of 95 and 5 wt%, respectively. TiO2 NRs in toluene were then added to the PDMS liquid prepolymer, and the solutions were stirred at 600 rpm for 10 min, in order to obtain a uniform and stable dispersion of NRs in PDMS. All solutions were left in the dark for several minutes to allow equilibration, and finally used to realize flat samples and microfluidic devices, according to a standard replica molding (REM) procedure33 (thermal curing at 70 °C for 20 min), starting from Si templates with a plane or structured surface, respectively. In particular, the Si master containing the microfluidic network was produced by photolithography and wet-etching, and was formed by four parallel capillaries of height (h) ∼3 μm, width (w) ∼20 μm and length (L) ∼1 cm, and by an integrated ruler fabricated side by side with the planar capillary, having parallel ticks separated by 50 μm, which allowed us to accurately measure the filling rate by optical microscopy. The final microfluidic devices were then realized by reversible sealing, mediated by conformal contact, of the nanocomposite replica to a glass substrate, which provided the fourth channel wall.
:1 base:curing agent) and TiO2 NRs in toluene were prepared at concentrations of 95 and 5 wt%, respectively. TiO2 NRs in toluene were then added to the PDMS liquid prepolymer, and the solutions were stirred at 600 rpm for 10 min, in order to obtain a uniform and stable dispersion of NRs in PDMS. All solutions were left in the dark for several minutes to allow equilibration, and finally used to realize flat samples and microfluidic devices, according to a standard replica molding (REM) procedure33 (thermal curing at 70 °C for 20 min), starting from Si templates with a plane or structured surface, respectively. In particular, the Si master containing the microfluidic network was produced by photolithography and wet-etching, and was formed by four parallel capillaries of height (h) ∼3 μm, width (w) ∼20 μm and length (L) ∼1 cm, and by an integrated ruler fabricated side by side with the planar capillary, having parallel ticks separated by 50 μm, which allowed us to accurately measure the filling rate by optical microscopy. The final microfluidic devices were then realized by reversible sealing, mediated by conformal contact, of the nanocomposite replica to a glass substrate, which provided the fourth channel wall.
Flat samples of bare PDMS and PDMS/TiO2 NRs nanocomposite were also produced by casting the materials on the surface of a flat Si substrate, followed by in situ thermal curing. These samples were used for morphological and chemical composition analysis.
Tests were also performed on nanocomposite samples prepared by using other two different concentrations of NRs (10 and 20 wt%), a different base:curing agent ratio (5:1) and different thermal curing parameters (140 °C for 10 min), but we decided to produce the final devices considering the parameters described above, since they gave the best conformal contact, the lowest surface roughness and the best fluidics results.
Photoirradiation and storage processes for reversible wettability changes
To enhance the hydrophilicity of the nanocomposite systems by exploiting the TiO2 NRs exposed on their surface, we irradiated both flat samples and microstructured PDMS/TiO2 replicas for 4 h by means of a pulsed Nd:YAG laser at 355 nm, (energy density of 7 mJ cm−2, repetition rate of 10 Hz, pulse duration of 4–6 ns). We achieved the complete recovery of the initial wettability of the samples by placing them in vacuum at a pressure of 3 × 10−3 mbar for 48 h. The pure elastomeric systems went through the same irradiation/vacuum treatment for comparison reasons.Characterization of samples
X-ray Photoelectron Spectroscopy (XPS) analysis was performed on flat samples to determine the chemical composition of the used surfaces. The measurements were performed in a SPECS XRC-1000 (SPECS LAB GmbH, Berlin) system equipped with two ultrahigh-vacuum (UHV) chambers, one for sample preparation and one for sample analysis. A Mg-Kα radiation source with energy of 1253.6 eV was employed. The charge-up of binding energy (BE) values was referenced to the C1 peak at 285.6 eV as an internal standard. The number of scans and the dimensions of the examined surfaces remained the same, so that the intensity of the obtained peaks could be directly compared.Atomic Force Microscopy (AFM) studies were performed on flat samples using a XE-100 PSIA instrument (Park Systems). The images were acquired in non-contact mode, working in air in a vibration-insulated environment (Table Stable TS-150). Single-beam silicon cantilevers coated with aluminum on the reflective side (type PPP-NCHR-10, Nanosensors) with typical elastic constant of 42 N m−1 and nominal tip radii of less than 10 nm were used. The drive frequency was ∼295 kHz, and the scan rate was between 0.1 and 0.5 Hz.
Fourier Transform Infrared (FT-IR) spectroscopy measurements in the 3700–3000 cm−1 spectral range were carried out on flat samples using a VERTEX 70 apparatus in transmittance mode at a resolution of 4 cm−1. To compensate for possible changes in the positioning of the samples, all the spectra were normalized to the Si–CH3 absorption at 1250 cm−1.
Measurements of the apparent WCA were carried out on flat samples with a KSVCAM200 instrument (KSV, Finland) on 15 different samples, every 30 min during the irradiation process and every 6 h during the vacuum storage. Distilled water was used as the liquid for these tests and was dispensed using a microsyringe (typical drop volume ∼1 μL). For each sample the WCA value was obtained as an average of 6 measurements recorded on different adjacent areas of the surface.
Scanning Electron Microscopy (SEM) analysis were performed on the microstructured samples by using a Nova NanoSEM 450 electron beam system (FEI Europe), operating with an acceleration voltage of 10 kV and an aperture size of 30 μm.
The study of water motion into the nanocomposite capillaries was conducted on the microstructured samples by releasing a 1 μL water drop at the edge of the microchannels, which were spontaneously filled by capillary action only after UV irradiation. The filling rate during the capillary rise was monitored by optical microscopy through a camera, using the lateral ruler integrated in the microfluidic system. The exact position of the fluid front along the microchannels during the entire filling process was determined using a dedicated viewer software with an acquisition rate of 30 frames × s−1. All the fluidics experiments were carried out within 24 h from the preparation of the systems, and the WCAs were found to be stable during this period.
Results and discussion
By introducing TiO2 nanorods into a PDMS matrix, we add a new functionality to conventional PDMS, namely the possibility of optically tuning its wetting properties. In detail, the developed nanocomposite material maintains the main characteristics of PDMS, in terms of applicability in conventional replica molding procedures and conformal contact on substrates of different nature, hence being suitable for the realization of microfluidic systems. Moreover, differently from standard PDMS molds, the produced PDMS/TiO2 nanocomposite samples can modulate their wetting characteristics using pulsed UV laser light irradiation, changing from hydrophobic to hydrophilic in a reversible manner.For the production of PDMS/TiO2 nanocomposites appropriate for microfluidic applications, the choice of the fabrication parameters is crucial, i.e. concentration of NRs to be embedded into the PDMS matrix, ratio of elastomer base (A) and curing agent (B), and thermal curing parameters. For this purpose, we first analyzed three different concentrations of NRs (5, 10, and 20 wt%) in a PDMS solution with A:B ratio equal to 10:1. We observed that 5 wt% of NRs in PDMS is the concentration that allows us to obtain highly reproducible patterned elastomeric materials, with a non-sticking behavior with the Si surface. In fact, by using a concentration of NRs of about 10 wt% the irreversible sealing of the elastomer on some regions of the Si template damages the features to be replicated, with consequent poor pattern replication. Moreover, when we prepared solutions of TiO2 NRs and PDMS with a concentration higher than 10 wt%, we observed, on one hand, a non-uniform mixing of the two parts with the occurrence of phase separation during the thermal curing of the elastomer, probably related to the high quantity of toluene involved in the process, and, on the other hand, high adhesion with the master surface. Concerning the ratio of PDMS base and curing agent, we tested 10:1 and 5:1 A:B with 5 wt% of NRs. We obtained the best conformal contact of the elastomeric structures onto different substrates (glass, silicon, and pure PDMS) by using 10:1 ratio. The increase of the quantity of curing agent with respect to that of the monomer induces a non-lasting contact of the structures, with liquid loss during the fluidic filling of the microchannels. The same effect was observed by using a curing temperature above 140 °C. Therefore, the optimized fabrication parameters that have to be considered in order to produce a nanocomposite elastomeric material suitable for soft lithographic procedures and microfluidics are the following: 5 wt% TiO2 in a PDMS solution characterized by a A:B ratio of 10:1, and cured at 70° for 20 min. Using these parameters, we first prepared flat samples of bare PDMS and PDMS/TiO2 NRs nanocomposite, in order to fully characterize them before and after UV pulsed laser irradiation. In particular, the surface chemical composition of both kinds of samples was examined through X-ray Photoelectron Spectroscopy (XPS) measurements. In Fig. 1, the O 1s peak at 530 eV, the C 1s peak at 285 eV, and the two components of the Si peak (the Si 2s peak at 154 eV and the Si 2p peak at 104 eV),34 all typical signals of PDMS, are visible both in the pure elastomer (black line) and in the nanocomposite (red line) samples. In addition, the spectrum associated with the nanocomposite sample shows peaks characteristic for the TiO2,35,36 namely the Ti 2p peak (which has two components, the Ti 2p1/2 at 465 eV and the Ti 2p3/2 at 460 eV) and a more pronounced O 1s peak at 530 eV. The existence of the TiO2 characteristic peaks demonstrates that there are NRs situated at the outer layer (∼10 nm in thickness) of the nanocomposite film, corresponding to the typical depth of analysis of the XPS system.
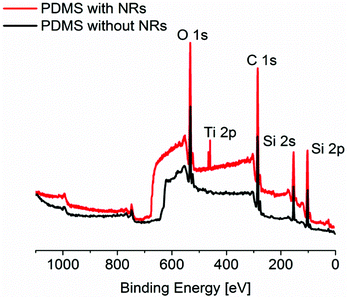 | ||
| Fig. 1 XPS survey Mg-Kα photoelectron spectra of PDMS with and without NRs, showing the main peaks of the two materials. | ||
After UV irradiation, performed by exploiting the conditions mentioned in the experimental part, the XPS analysis shows no changes in the spectra of the pure PDMS films, whereas it reveals UV-driven modifications in the surface chemistry of the PDMS/TiO2 nanocomposite films, as demonstrated by the O1s peak. In particular, the main components of the deconvoluted O1s peaks of non-irradiated and irradiated nanocomposite films are displayed in Fig. 2a and 2b, respectively.
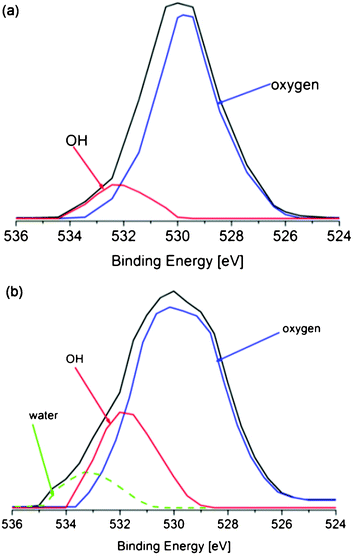 | ||
| Fig. 2 XPS resolved spectra of the O 1s region for PDMS/TiO2 samples before (a) and after (b) UV irradiation, showing the main components of the peak. | ||
As shown, for the non-irradiated PDMS/TiO2 samples (Fig. 2a), two resolved peaks at 530 and 532 eV evolved. The most intense peak at 530 eV is attributed to the oxygen present in the PDMS molecules34 and to the Ti–O bonds in the NRs,35 whereas a small contribution of the hydroxyl groups, which contaminate the NRs facets already before UV irradiation, is shown at 532 eV.37 After UV irradiation, the O1s peak of the nanocomposite samples is deconvoluted in three different contributions, at 530, 532 and 533.5 eV (Fig. 2b). The peak at 532 eV clearly shows a higher distribution than in the non-irradiated sample, demonstrating the existence of an increased number of OH groups on the surface of the nanocomposites after UV irradiation. Importantly, the presence of the extra peak at 533.5 eV is due to molecular H2O adsorbed on the NRs surface after the UV irradiation.38,39 Since the peaks associated with the increased number of hydroxyl groups and water are detected exclusively in the UV-irradiated PDMS/TiO2 NRs samples and not in the pure PDMS, it can be safely assumed that after irradiation they get adsorbed on the TiO2 NRs.
The introduction of OH species in the PDMS/TiO2 nanocomposite samples after UV irradiation is confirmed by Fourier Transform Infrared (FT-IR) analysis performed on flat elastomer and nanocomposite samples. The measurements on pure PDMS reveal no changes before and after UV irradiation (see Supporting Information, Figure S1). On the contrary, the OH band (3100–3500 cm−1) in the nanocomposites presents remarkable changes before and after UV irradiation (Fig. 3). Particularly, the signals from the dissociatively and molecularly physisorbed H2O (ν(OH) at ∼3300 cm−1 and ν(H2O) at ∼3200 cm−1, respectively) are substantially enhanced, suggesting an increase in the amount of H2O on the NRs facets. FT-IR analysis was also performed on the flat nanocomposite samples after vacuum storage, highlighting a reduction of the intensity of the OH-related peak with the recovery of the initial low hydroxylation degree of the samples. Therefore, the OH species introduced to the surface upon UV photoirradiation are metastable and they disappear after two days of vacuum treatment.
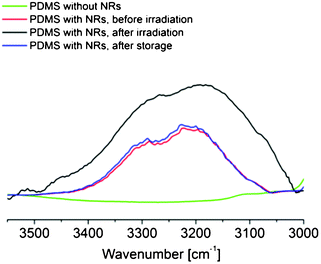 | ||
| Fig. 3 FT-IR spectra of PDMS/TiO2 films throughout a cycle of the UV irradiation and vacuum storage process in the 3600–3000 cm−1 range. | ||
A widely accepted mechanism proposed in the literature to explain the effect of UV light on the TiO2 is in full agreement with our findings. Briefly, UV excitation generates holes on the TiO2 surface, which lead to the formation of oxygen vacancies. Ambient water molecules coordinate dissociatively or by physisorption to the formed oxygen vacancies, generating an OH-rich surface.37 Subsequent storage in ambient conditions, or vacuum storage for a faster procedure, induces detachment of the OH groups implanted onto the surface and their replacement by atmospheric oxygen as soon as the samples come in contact with the ambient atmosphere.37,40–43 Previous work by our group have demonstrated that under the UV pulsed laser irradiation conditions used in this work the photocatalytic effect of TiO2 is inhibited13,37,40,41 whereas, on the contrary, is favored by the use of UV lamps.26 Hence, the reversible hydroxylation of the PDMS/TiO2 samples is the only effect induced from the UV irradiation-vacuum storage treatment.
Since the hydroxyl groups and water adsorbed on the TiO2 NRs surface after UV irradiation (found by XPS and FTIR measurements) originate from the ambient atmosphere, we can safely assume that the NRs are exposed on the surface of the composite elastomeric films, and are in contact with ambient humidity. The actual presence of the NRs on the nanocomposite surface is further supported by Atomic Force Microscope (AFM) phase images (see Supporting Information, Figure S2†). The surfaces of the nanocomposite films are clearly composed of different phases, indicating different materials (elastomer and NRs) in contrast to the pure elastomer samples, where only one phase is detected.
Furthermore, we examined the effect of the presence of the TiO2 NRs on the wetting surface properties of the flat nanocomposite systems by performing WCA measurements throughout the UV irradiation and vacuum storage procedures. Fig. 4a illustrates the static WCA values measured on flat elastomeric surfaces with and without NRs at scheduled time intervals during the irradiation and the vacuum storage processes. The WCA of the pure PDMS remains stable at 116 ± 2° before, during and after the irradiation and the storage processes, confirming that the TiO2 fillers are exclusively responsible for any wettability differences induced by light. In the case of the PDMS/TiO2 nanocomposites the initial WCA is 105 ± 2°, slightly lower than that measured on pure elastomer samples. The decreased WCA values of the nanocomposite samples can be attributed to the increased surface hydrophilicity due to the TiO2 NRs present on the sample surface (independent WCA measurements on films of pure TiO2 NRs deposited by drop casting on glass or silicon substrates gave an average WCA value of 72 ± 2°). The UV irradiation of the nanocomposites causes a decrease in the WCA of about ∼20°, leading to a contact angle of 84 ± 2°. This decrement is attributed to the UV-induced adsorption of hydroxyl groups and water on the TiO2 NRs, previously proved by FT-IR and XPS measurements, which are expected to increase the hydrophilization of the samples. In order to preserve the characteristics of the PDMS matrix, we irradiated the samples for four hours. In fact, although the increase of the irradiation time to five hours leads to a lower WCA (77 ± 2°), signs of degradation of the PDMS matrix were observed, such as loss of transparency and poor conformal contact on substrates. This compromises the reproducibility of the technique and the possibility of using the nanocomposite samples for the production of functional microfluidic devices. A reduction of irradiation time is possible using a laser system characterized by high repetition rate or different pulse duration. Finally, after a short vacuum storage period, the PDMS/TiO2 samples recover their initial hydrophobic character. This result is clearly related to the FT-IR findings that after the vacuum treatment of the PDMS/TiO2 NRs samples they recover their initial low hydroxylation degree. The same result can be obtained by storing the samples for about four days under laboratory or ambient light conditions. The reversible wetting cycles were repeated many times on various nanocomposite films, without any apparent fatigue (Fig. 4b).
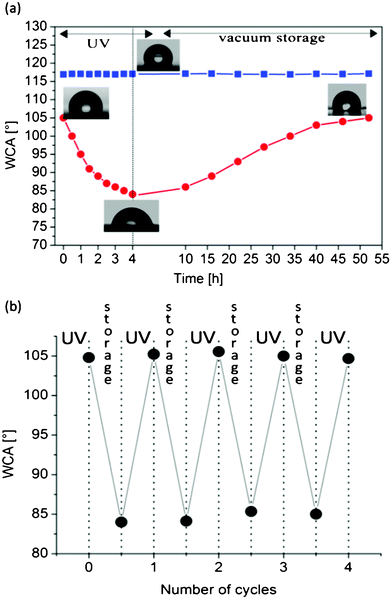 | ||
| Fig. 4 (a) WCA measurements performed on PDMS/TiO2 NRs samples during a cycle of UV irradiation and vacuum/storage (•) and of PDMS samples without NRs during the same UV-irradiation/storage cycle (■). The thickness of each point represents the experimental error of the measurements. Pictures of the water drops lying on the nanocomposite surface are shown after their preparation, at the end of the irradiation process and after successive storage in vacuum conditions. (b) Reversible wettability changes during cyclic alternations of UV illumination and vacuum storage. | ||
The reason for using TiO2 NRs instead of spherical nanoparticles (NPs) in this work is to maximize the hydrophilicization efficiency of the TiO2 nanofillers. Actually, the surface to volume ratio of the rods is (2/R)+(2/L), where R is the radius of the NRs and L their length, whereas the one for the spherical NPs of radius r is 3/r. For the NRs used in this work (R = 1.5 nm and L = 20 nm) the surface to volume ratio is 1.43. This ratio is bigger than the one calculated for NPs with spherical shape when their diameter becomes greater than 4.18 nm, which is the case for the vast variety of the reported chemically synthesized TiO2 NPs.44–47 Therefore, the TiO2 NRs have increased effectiveness with respect to the NPs in the adsorption of hydroxyl groups after UV irradiation, which are responsible for their UV light induced increased wettability. Nevertheless, the increased hydrophilicization efficiency of the TiO2 NRs could be somehow compromised by the formation of agglomerates in the PDMS matrix.
We exploited the hydrophobic to hydrophilic switch of the developed nanocomposites in order to produce fluidic systems with photoinduced spontaneous liquid flow in their microchannels, as described in the experimental section. The produced microfluidic devices were first investigated by SEM. As presented in Fig. 5a, the microchannels and the integrated ruler are well replicated by the developed nanocomposite, without the presence of distortions or breaking in the features. Moreover, the higher magnification view (Fig. 5b) demonstrates the uniform dispersion of the NRs, without evident aggregates along the microchannels or onto the surface of the interchannel separation feature. In this way, the conformal contact of replica onto the substrate is favored, preventing water losses during the filling process.
 | ||
| Fig. 5 SEM planar views of the produced PDMS/TiO2 microchannels: (a) four parallel capillaries and the integrated ruler on the right side; (b) high magnification image of the two central microchannels. | ||
Water flow tests were performed on microfluidic channels of the same geometry made of PDMS/TiO2 NRs nanocomposites and of bare PDMS as reference. As shown in Fig. 6a, the structured surface of the PDMS/TiO2 mold is directly exposed to a homogeneous UV laser beam, in order to achieve a uniform irradiation of all the walls of the microchannels. Fig. 6b shows that water penetration is completely forbidden in pure PDMS and in PDMS/TiO2 microchannels before UV irradiation, due to the hydrophobic nature of the involved surfaces (see Fig. 4). After UV irradiation, water spontaneously penetrates only in the PDMS/TiO2 microchannels (Fig. 6c), since the light-induced wettability changes make the channels walls hydrophilic, as previously demonstrated by the WCA measurements presented in Fig. 4a.
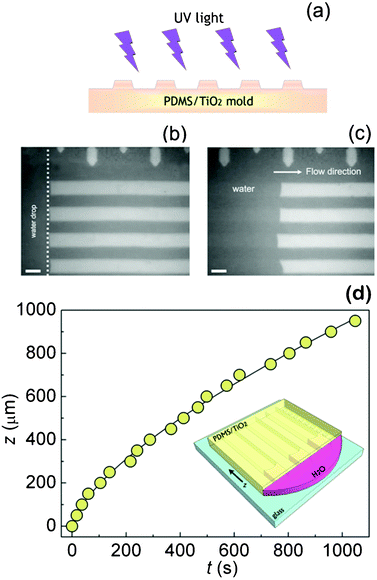 | ||
| Fig. 6 (a) Schematic representation of the irradiation process. Optical microscope images (b) of a water droplet released at the edges of a non-irradiated PDMS/TiO2 nanocomposite capillaries, into which it cannot penetrate, and (c) of water filling the irradiated microchannels. The length of the marker is 20 μm in both panels. (d) Experimental z(t) data (circles) for water rising into the PDMS/TiO2 microchannels irradiated with UV light. The superimposed line is a guide for the eye only. The inset shows a schematic representation of the water filling experiment performed on a nanocomposite network of capillaries (features not to scale). | ||
The coordinate z of the liquid front along the flow direction in the nanocomposite microchannels is strictly dependent on the involved surface tensions and the effective contact angle. The experimental data providing the z coordinate in relation with time (z, t) for the UV irradiated microfluidic nanocomposite channels were collected by real-time observation through an optical microscopy camera and the obtained experimental points are presented in Fig. 6d. We estimated that the average velocity of water in the microchannels is about 0.9 μm s−1, which means that water rinses 930 μm of the capillary length in about 1000 s. The effective contact angle observed within the microchannel is below 90° (Fig. 6c), in agreement with the observation of spontaneous capillary penetration phenomena. In particular, we point out that the found meniscus deviation from a usual Poiseuille parabolic profile is likely to be related to the surface roughness of the capillary walls and to the free energy conditions, which may be dishomogeneous at sub-μm scale, and can determine a consequent enhancement of the energy dissipation along the contact line.48 Generally speaking, the steady state flow of a Newtonian fluid (like water) inside capillaries as described by the Poiseuille law would ultimately lead to a Washburn penetration behavior on the nanocomposite surface.49
However, in our system, we found a complex penetration behavior with local discontinuities in the velocity temporal dependence, because of the pinning-depinning behavior associated to the nanoscale surface roughness (according to AFM measurements (results not shown), pure elastomer samples exhibit a lower surface roughness (0.98 ± 0.20 nm) compared to nanocomposite samples (22.66 ± 0.20 nm)) or local chemical discontinuities induced by the exposed TiO2 NRs. In particular, as the contact line moves, the interaction with the local wall topology plays a crucial role in the wetting process,50 and the pinning-depinning effects lead to a stick slip motion of the meniscus at the solid/liquid interface. Hence, the wetting front advances on the solid surface with friction properties dependent on the wall roughness,51 that in turn influences the energy balance of the system and consequently the dynamics of the capillary flow.52 Finally, we notice that, after the two days storage period in vacuum, the PDMS/TiO2 NRs nanocomposite capillaries fully recovered their hydrophobic character, exactly as in the case of the flat nanocomposite films presented in Fig. 4a, and the flow of the water in the microchannels is again forbidden.
We demonstrated the spontaneous penetration of water in irradiated PDMS/TiO2 microchannels exploiting the control over the wetting properties of the capillaries walls. Our approach can also offer the possibility of locally tuning the wetting properties of the microchannels, with application in generation of multiple emulsions and Laplace valves. In fact, in microfluidic devices exploited for multiple emulsions, the wettability of a channel influences the superficial properties of the drop makers, and it is crucial for the regulation of the drop formation.53 Moreover, for Laplace valves, the proper spatial control of the wettability of the microchannels favors the tuning of the Laplace pressure barrier in a desired position, hence controlling fluid flow.54
Conclusions
We developed hydrophobic nanocomposites of PDMS/TiO2 NRs, which upon pulsed UV laser irradiation swap to hydrophilic materials, due to hydroxyl groups and molecular water adsorbed on the NRs exposed on the surface of the nanocomposites. By exploiting these materials, we realized microfluidic systems of specific geometries, which allow the spontaneous penetration of water into the microchannels only after UV-irradiation, due to the improved hydrophilicity of their walls. In contrast, water penetration is prohibited into bare PDMS microchannels both before and after UV irradiation. The nanocomposite systems show a reversible change in their wettability and recover their hydrophobic character upon vacuum storage, making the capillaries again forbidden to water. In order to perform the UV treatment of the presented systems, it was possible to use the last generation of compact pulsed lasers or LEDs, thus simplifying the experimental set-up and reducing the cost of the procedure. The successful and easy realization of microfluidic chips with tunable surface properties and optically controlled liquid motion, using versatile nanoparticles, opens the way to use such devices in applications requiring the controlled transport of molecules in aqueous environments.55References
- C. D. Bain, G. D. Burnett Hall and R. R. Montgomerie, Nature, 1994, 372, 414 CrossRef CAS.
- B. Gallardo, V. K. Gupta, F. D. Eagerton, L. I. Jong, V. S. Craig, R. R. Shah and N. L. Abbott, Science, 1999, 283, 57 CrossRef CAS.
- X. Pei, A. Fernandes, B. Mathy, X. Laloyaux, B. Nysten, O. Riant and A. M. Jonas, Langmuir, 2011, 27, 9403 CAS.
- N. Nishikawa, A. Uyama, T. Kamitanaka, H. Mayama, Y. Kojima, S. Yokojima, S. Nakamura, K. Tsujii and K. Uchida, Chemistry–An Asian Journal, 2011, 6, 2400 CrossRef CAS.
- A. Athanassiou, M. I. Lygeraki, D. Pisignano, K. Lakiotaki, M. Varda, E. Mele, C. Fotakis, R. Cingolani and S. H. Anastasiadis, Langmuir, 2006, 22, 2329 CrossRef CAS.
- A. Athanassiou, M. Varda, E. Mele, M. I. Lygeraki, D. Pisignano, M. Farsari, C. Fotakis, R. Cingolani and S. H. Anastasiadis, Appl. Phys. A, 2006, 83, 351 CrossRef CAS.
- M. I. Lygeraki, E. Tsiranidou, S. H. Anastasiadis, C. Fotakis, D. Pisignano, R. Cingolani and A. Athanassiou, Appl. Phys. A, 2008, 91, 397 CrossRef CAS.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Nature, 1997, 388, 431 CrossRef CAS.
- M. Miyauchi, A. Nakajima, T. Watanabe and K. Hashimoto, Chem. Mater., 2002, 14, 2812 CrossRef CAS.
- S. N. Das, J. H. Choi, J. P. Kar and J. M. Myoung, Appl. Surf. Sci., 2009, 255, 7319 CrossRef CAS.
- X. Feng, J. Zhai and L. Jiang, Angew. Chem., Int. Ed., 2005, 44, 5115 CrossRef CAS.
- G. Kwak, M. Lee and K. Yong, Langmuir, 2010, 26, 9964 CrossRef CAS.
- G. Caputo, B. Cortese, C. Nobile, M. Salerno, R. Cingolani, G. Gigli, P. D. Cozzoli and A. Athanassiou, Adv. Funct. Mater., 2009, 19, 1149 CrossRef CAS.
- K. Ichimura, S.-K. Oh and M. Nakagawa, Science, 2000, 288, 1624 CrossRef CAS.
- S.-K. Oh, M. Nakagawa and K. Ichimura, J. Mater. Chem., 2002, 12, 2262 RSC.
- F. Villafiorita-Monteleone, G. Caputo, C. Canale, P. D. Cozzoli, R. Cingolani, D. Fragouli and A. Athanassiou, Langmuir, 2010, 26, 18557 CrossRef CAS.
- A. M. Christensen, D. Chang-Yen and B. K. Gale, J. Micromech. Microeng., 2005, 15, 928 CrossRef.
- S. R. Sershen, G. A. Mensing, N. Ng, N. J. Halas and D. J. Beebe, Adv. Mater., 2005, 17, 1366 CrossRef CAS.
- R. H. Farahi, A. Passian, T. L. Ferrel and T. Thundat, Appl. Phys. Lett., 2004, 85, 4237 CrossRef CAS.
- M. K. Chaudhury and G. Whitesides, Science, 1992, 256, 1539 CAS.
- D. Erickson and D. Q. Li, Anal. Chem. Acta, 2004, 507, 11 CrossRef CAS.
- L. Caprioli, E. Mele, F. E. Angilè, S. Girardo, A. Athanassiou, A. Camposeo, R. Cingolani and D. Pisignano, Appl. Phys. Lett., 2007, 91, 113113 CrossRef.
- G. M. Whitesides and A. D. Stroock, Phys. Today, 2001, 54, 42 CrossRef CAS.
- K. Nakata, H. Kimura, M. Sakai, T. Ochiai, H. Sakai, T. Murakami, M. Abe and A. Fujishima, Appl. Mater. Interfaces, 2010, 2, 2485 CrossRef CAS.
- H. Makamba, J. H. Kim, K. Lim, N. Park and J. H. Hahn, Electrophoresis, 2003, 24, 3607 CrossRef CAS.
- H. Nagai, T. Irie, J. Takahashi and S.-I. Wakida, Biosens. Bioelectr., 2007, 22, 1968 CrossRef CAS.
- Q. Zhang, J. J. Xu, Y. Liu and H. Y. Chen, Lab Chip, 2008, 8, 352 RSC.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563 CrossRef CAS.
- S. M. Stavis, J. B. Edel, K. T. Samiee and H. G. Craighead, Lab Chip, 2005, 5, 337 RSC.
- Y. C. Tan, J.-S. Fisher, A. I. Lee, V. Cristini and A. P. Lee, Lab Chip, 2004, 4, 292 RSC.
- C. L. Walsh, B. M. Babin, R. W. Kasinskas, J. A. Foster, M. J. McGarry and N. S. Forbes, Lab Chip, 2009, 9, 545 RSC.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem. Int. Ed., 1998, 37, 550 CrossRef CAS.
- X. Deng, R. Luo, H. Chen, B. Liu, Y. Feng and Y. Sun, Colloid Polym. Sci., 2007, 285, 923 CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664 CrossRef CAS.
- E. McCafferty and J. P. Wightman, Surf. Interface Anal., 1998, 26, 549 CrossRef CAS.
- G. Caputo, C. Nobile, T. Kipp, L. Blasi, V. Grillo, E. Carlino, L. Manna, R. Cingolani, P. D. Cozzoli and A. Athanassiou, J. Phys. Chem. C, 2008, 112, 701 CAS.
- G. W. Simmons and B. C. Beard, J. Phys. Chem., 1987, 91, 1143 CrossRef CAS.
- J. Pouilleau, D. Devilliers and H. Groult, J Mater Sci., 1997, 32, 5645 CrossRef CAS.
- G. Caputo, C. Nobile, R. Buonsanti, T. Kipp, L. Manna, R. Cingolani, P. D. Cozzoli and A. Athanassiou, J. Mater. Sci., 2008, 43, 3474 CrossRef CAS.
- G. Caputo, R. Cingolani, P. D. Cozzoli and A. Athanassiou, Phys. Chem. Chem. Phys., 2009, 11, 3692 RSC.
- Z. Zhang, C. C. Wang, R. Zakaria and J. Y. Ying, J. Phys. Chem. B, 1998, 102, 10871 CrossRef CAS.
- C.-Y. Wang, H. Groenzin and M. J. Shultz, Langmuir, 2003, 19, 7330 CrossRef CAS.
- T. K. Misra and C.-Y. Liu, J. Colloid Interface Sci., 2007, 310, 178 CrossRef CAS.
- G. W. Lee and S. M. Choi, in Eco-Materials Processing and Design VIII, ed. H. Kim, J. Hojo and S. W. Lee, Materials Science Forum 2007, pp. 544–545 Search PubMed.
- H. D. Jang, S.-K. Kim and S.-J. Kim, J. Nanopart. Res., 2001, 3, 141 CrossRef CAS.
- L. Zhao and J. G. Yu, J. Colloid Interface Sci., 2006, 304, 84 CrossRef CAS.
- H. Kusumaatmaja, C. M. Pooley, S. Girardo, D. Pisignano and J. M. Yeomans, Phys. Rev. E, 2008, 77, 067301 CrossRef CAS.
- E. W. Washburn, Phys. Rev., 1921, 17, 273 CrossRef.
- E. Rio, A. Daerr, B. Andreotti and L. Limat, Phys. Rev. Lett., 2005, 94, 0245031 CrossRef.
- M. K. Stukan, P. Ligneul, J. P. Crawshaw and E. S. Boek, Langmuir, 2010, 26, 13342 CrossRef CAS.
- S. Girardo, S. Palpacelli, A. De Maio, R. Cingolani, S. Succi and D. Pisignano, Langmuir, 2012, 28, 2596 CrossRef CAS.
- A. R. Abate, J. Thiele, M. Weinhart and D. A. Weitz, Lab Chip, 2010, 10, 1774 RSC.
- G. Takei, M. Nonogi, A. Hibara, T. Kitamori and H.-B. Kim, Lab Chip, 2007, 7, 596–602 RSC.
- N. Sakai, R. Wang, A. Fujishima, T. Watanabe and K. Hashimoto, Langmuir, 1998, 14, 5918 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: FT-IR spectra of nanocomposite samples before and after irradiation and after storage; AFM phase images of pure PDMS elastomer and PDMS/TiO2 NRs nanocomposite samples See DOI: 10.1039/c2ra20573d/ |
| This journal is © The Royal Society of Chemistry 2012 |