Structure modulation, argentophilic interactions and photoluminescence properties of silver(I) coordination polymers with isomeric N-donor ligands†
Kamal
Kumar Bisht
a,
Amal
Cherian Kathalikkattil
ab and
Eringathodi
Suresh
*a
aAnalytical Discipline and Centralized Instrument Facility, Central Salt and Marine Chemicals Research Institute, Council of Scientific and Industrial Research (CSIR) G.B.Marg, Bhavnagar, 364 002, Gujarat, India. E-mail: esuresh@csmcri.org; sureshe123@rediffmail.com
bDepartment of Chemical Engineering, Pusan National University, Jangjun-Dong, Gumjung-gu, Busan, 609-735, Republic of Korea
First published on 13th July 2012
Abstract
Synthesis, crystal structure, and photoluminescence studies of a one dimensional silver coordination polymer {[Ag(L1)(CH3CN)][Ag(L1)(OTf)](OTf)}n (1) comprising of Ag(I) and a new flexible organic linker bis(pyridine-3-ylmethyl)terephthalate (L1) exhibiting closed-shell d10⋯d10 argentophilic interaction, is reported. The compound 1 consists of two kinds of (double and single stranded) 1D coordination polymeric chains running parallel and displaying short Ag(I)⋯Ag(I) distances. Notably, the coordinated triflate anion, μ2-bridging mode of acetonitrile and stacking interactions play an important role in the realization of 1D quadruple metal–organic chain in compound 1. Treatment of silver triflate with isomeric ligand, bis(pyridine-2-ylmethyl)terephthalate (L2) resulted in a polymorph of previously reported 1D zigzag coordination polymer {[Ag(L2)](OTf)(H2O)}n (2). Both organic ligands and Ag coordination polymers are characterized by various analytical techniques. Crystal and molecular structures of the isomeric ligands (L1, L2) and their Ag(I) coordination polymeric architectures (1, 2) are correlated with their photoluminescence properties.
Introduction
Research in the field of coordination polymers over the past two decades has devised a number of crystal engineering approaches towards the design and synthesis of metal organic frameworks or coordination polymeric materials with different dimensionality and network topology resulting in applications in material chemistry.1 In the case of 1D coordination polymers, besides their aesthetic appeal and ease of synthesis, their potential applications in various fields such as photoluminescence, molecular wires, catalysis, and ion encapsulation offer a driving force for researchers working in the area of material science.2 In general, coordination polymers are synthesized by self-assembly processes between metal ions with versatile coordination geometry and multitopic organic linkers of different flexibility.3 Ag(I) ion with a d10 electronic configuration and adaptable coordination geometry can accommodate a variety of ligands, generating multidimensional networks in an unpredictable fashion.4 Such self assemblies can be directed by templating agents such as solvent molecules.3k Huang and co-workers have shown the template effect of acetonitrile solvent in the assembly of a laminar silver coordination polymer.4j Luminescence is one of the interesting features associated with silver coordination architectures that fascinates researchers due to its probable practical implications.5 Previous and many recent reports indicate that the short Ag⋯Ag separations, popularly known as argentophilic interactions play a crucial role in the manifestation of the luminescence properties of silver coordination polymers in addition to other factors such as π⋯π stacking.6 Recently, Munakata et al., have shown that π⋯π stacking of the ligand moiety in silver coordination polymers can result in the red shift of emission wavelengths as compared to those of the corresponding ligands.7We have been involved in investigating various molecular interactions in a variety of supramolecular systems including coordination polymers and hybrid organic materials.8 Herein, we report structural studies of two isomeric ligands (L1, L2) and their one dimensional Ag(I) coordination polymers {[Ag(L1)(CH3CN)][Ag(L1)(OTf)](OTf)}n (1) and {[Ag(L2)](OTf)(H2O)}n (2). Both coordination polymers 1 and 2 exhibit luminescence in solid and solution states at room temperature. Specifically, compound 1 exhibits blue emission with a remarkable red shift in the solid state; however, emission profiles of both complexes in acetonitrile solution and that of 2 in the solid state are comparable with their respective organic linkers. Single crystal structural analyses reveal short Ag⋯Ag separations in the coordination polymer 1. The luminescence behaviour of coordination polymers 1 and 2 is correlated with the solid state structural features.
Experimental
Materials
All reagents for ligand synthesis and silver triflate were purchased from Sigma–Aldrich. Solvents used for ligand synthesis and complexation were obtained from SD Fine Chemicals, India. All the reagents and solvents were used as received without further purification. L1 and L2 were prepared following a known procedure.13Physical measurements
CHN analyses were carried out using a Perkin–Elmer 2400 CHNS/O analyzer. IR spectra were recorded using KBr pellets on a Perkin–Elmer GX FTIR spectrometer. For each IR spectra 10 scans were recorded at 4 cm−1 resolution. 1H and 13C NMR spectra for the compounds were recorded on Bruker AX 500 spectrometer (500 MHz or 200 MHz) at temperature 25 °C. NMR Spectra was calibrated with respect to internal reference TMS. TGA analyses were carried out using METTLER TOLEDO STARe SW 8.10, luminescence spectra and UV–vis spectra were recorded using a Fluorolog Horiba Jobin Yvon spectrophotometer and Shimadzu UV-3101PC spectrophotometer respectively. X-Ray powder diffraction data were collected using a Philips X-Pert MPD system with Cu-Kα radiation.X-Ray crystallography
Summary of crystallographic data and details of data collection for L1, L2, complex 1 and complex 2 are given in Table S1.† Single crystals with suitable dimensions were chosen under an optical microscope and mounted on a glass fiber for data collection. Intensity data for all the four crystals were collected using Mo-Kα (λ = 0.71073 Å) radiation on a Bruker SMART APEX diffractometer equipped with CCD area detector at 110 K for L1, 1 and 2, and 293 K for L2. The data integration and reduction were processed with SAINT software.9 An empirical absorption correction was applied to the collected reflections with SADABS.10 The structures were solved by direct methods using SHELXTL and were refined on F2 by the full-matrix least-squares technique using the program SHELXL-97.11,12 All non-hydrogen atoms were refined anisotropically till convergence was reached. Hydrogen atoms attached to the organic moieties are either located from the difference Fourier map or stereochemically fixed in all the compounds. In the case of Complex 1, C43 carbon atom of the coordinated acetonitrile molecule (with Ag2) showed slightly higher temperature factor. Hence C43 is refined only isotropically in the final cycles of refinement.Synthesis of bis(pyridine-3-ylmethyl)terephthalate (L1) and bis(pyridine-2-ylmethyl)terephthalate (L2)
Ligands L1 and L2 were synthesized under a nitrogen atmosphere. Pyridine-3-methanol or pyridine-2-methanol (2.25 g, 20 mmol) and 50 ml dichloromethane were taken in a round bottomed flask (capacity 250 ml) and placed in an ice-bath. After 15 min triethylamine (3 ml, 20 mmol) was added with continuous stirring. The reaction was allowed to reach room temperature after dropwise addition of 70 ml dichloromethane solution of terephthaloyl chloride (2 g, 10 mmol). The reaction mixture was further allowed to stir at room temperature for 8 h then washed thrice with water (3 × 20 ml) and dried over Na2SO4. Finally, organic solvent was removed by rotary evaporator to give L1 (or L2) as an off white powder in good yield (∼2.4 g, 68%) that was recrystallized from acetonitrile. The synthetic procedure for L1 is depicted as scheme S2 in the ESI.†L1: yield: 68%. CHN Data for C20H16O4N2: Calc (%): C, 68.96; H, 4.63; N, 8.04; Observed (%): C, 68.63; H, 4.67; N, 7.87. IR (KBr, νmax, cm−1): 2967(m), 1716(s), 1581(m), 1504(w) 1453(s), 1271(s, b), 1104(s, b), 1021(m), 924(m), 828(m), 796(s), 726 (s), 708(s). 1H NMR (CD3CN, 200 MHz): δ 5.38 (s, 4H, CH2), 7.38 (q, 2H, pyridine), 7.86 (d, 2H, pyridine), 8.11 (s, 4H, aromatic), 8.57 (d, 2H, pyridine), 8.70 (s, 2H, pyridine). L2: yield: 64%. CHN Data for C20H16O4N2: Calc (%): C, 68.96; H, 4.63; N, 8.04; Observed (%): C, 68.72; H, 4.66; N, 7.92. IR (KBr, νmax, cm−1): 2926(w), 1722(s), 1584(m), 1503(w) 1439(m), 1270(s, b), 1117(s, b), 1009(m), 988(m), 876(m), 761(s), 727 (s), 588(w). 1H NMR (CD3CN, 200 MHz): δ 5.44 (s, 4H, CH2), 7.31 (q, 2H, pyridine), 7.50 (d, 2H, pyridine), 7.80 (dt, 2H, pyridine), 8.16 (s, 4H, aromatic), 8.57 (m, 2H, pyridine).Synthesis of 1 and 2
Ligand L1 or L2 (350 mg, 1 mmol) and silver triflate (257 mg, 1 mmol) were dissolved in 25 mL acetonitrile with continuous stirring at ambient temperature. The reaction mixture was allowed to stir for 6 h in the dark and then filtered. The clear yellowish filtrate was kept in the dark for crystallization. The X-Ray quality crystals of coordination polymer 1 (or 2) obtained within a week along with little micro/poly crystalline material that was soluble in common organic solvents such as acetonitrile and dimethylformamide. Phase pure compounds 1 and 2 were obtained upon recrystallization of micro/poly crystalline compound from acetonitrile solutions. Complex1: yield: 88%. CHN Data for C44H35Ag2F2N5O14S2: Calc (%): C, 42.22; H, 2.82; N, 5.60; Observed (%): C, 41.93; H, 2.97; N, 5.58. IR (KBr, νmax, cm−1): 2373(w), 1720(s), 1606(m), 1505(w) 1444(m), 1272(s, b), 1108(s, b), 1027(s), 892(w), 800(m), 728(s), 632(s), 514(m). 1H NMR (CD3CN, 200 MHz): δ 5.39 (s, 4H, CH2), 7.43 (q, 2H, pyridine), 7.91 (d, 2H, pyridine), 8.12 (s, 4H, aromatic), 8.55 (d, 2H, pyridine), 8.67 (m, 2H, pyridine). Complex2: yield: 86%. CHN Data for C21H18AgF3N2O8S: Calc (%): C, 40.47; H, 2.91; N, 4.49; Observed (%): C, 40.45; H, 2.96; N, 4.46. IR (KBr, νmax, cm−1): 3473(b), 1715(s), 1603(m), 1575(w) 1447(s), 1272(s, b), 1160(s), 1118(s, b), 1029(s), 876(m), 766(s), 724(s), 638(m) 512(w). 1H NMR (CD3CN, 200 MHz): δ 5.42 (s, 4H, CH2), 7.40 (t, 2H, pyridine), 7.58 (d, 2H, pyridine), 7.89 (t, 2H, pyridine), 8.09 (s, 4H, aromatic), 8.57 (d, 2H, pyridine).Results and discussion
Ag(I) having d10 configuration is a soft-metal ion often producing versatile complex geometries dictated more by the conformational flexibility of the ligands than the geometric demands of the metal center. In our attempt to investigate the design and control of the self-assembly of organic/inorganic supramolecular architectures with flexible isomeric angular dipyridyl ligands (L1 and L2) two new Ag(I) coordination polymers 1 and 2 have been synthesized and their luminescence behaviour has been correlated with structural features revealed from single crystal X-ray diffraction technique. Interestingly, the coordination polymer 1 fluoresces blue radiation at room temperature and comprise of two types of molecular chains in which four silver ions are in close proximity, with Ag⋯Ag contacts ranging from 3.3 to 3.7 Å resulting in infinite quadruple assemblies. A simpler supramolecular 1D coordination polymer {[Ag(L2)](OTf)(H2O)}n (2) resulted upon treatment of silver triflate with isomeric ligand L2.All four compounds L1, L2, 1 and 2 were thoroughly characterized by various physico-chemical techniques including single crystal X-ray diffraction. The spectroscopic data of 1 and 2 are very similar. IR spectra of all four compounds are given in the ESI† as S3–6. High intensity bands at 1722 cm−1 and 1716 cm−1 for asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes that envelop symmetric bands of terephthalate moiety are prominent features of the IR profiles of ligands L1 and L2 respectively. The IR spectra of coordination polymers 1 and 2 show two strong bands at 1272, 1027 cm−1 and 1272, 1029 cm−1 respectively that can be assigned to the triflate anions. Absorptions bands appearing at 1720 cm−1 for 1 and at 1715 and 1669 cm−1 for 2 are accredited to the carbonyl stretching of the ligands (ester). Additionally the broad band near 3473 cm−1 in 2 is attributable to the lattice water.
O stretching modes that envelop symmetric bands of terephthalate moiety are prominent features of the IR profiles of ligands L1 and L2 respectively. The IR spectra of coordination polymers 1 and 2 show two strong bands at 1272, 1027 cm−1 and 1272, 1029 cm−1 respectively that can be assigned to the triflate anions. Absorptions bands appearing at 1720 cm−1 for 1 and at 1715 and 1669 cm−1 for 2 are accredited to the carbonyl stretching of the ligands (ester). Additionally the broad band near 3473 cm−1 in 2 is attributable to the lattice water.
The 1H NMR spectrum of all four compounds (L1, L2, 1 and 2) were recorded in acetonitrile–d3 solutions. Both ligands L1 and L2 show six well distinguished sets of resonance signals in the chemical shift range δ = 5.3–8.7, confirming their symmetric structures (ESI†S7–11). 1H NMR spectra of complexes 1 and 2 show close resemblance with their respective ligands. Both complexes are air-stable and retain their crystalline integrity for several months under ambient conditions.
Thermogravimetric analysis (ESI†S12) was performed to gauge the thermal stability, desolvation and decomposition pattern for 1 and 2. Lattice water molecule present in 2 expels (calculated weight loss = 2.89% and observed weight loss = 3.07%) in the temperature range 120–210 °C and both complexes exhibit excellent thermal stability up to 250 °C.
The homogeneity of the synthesized coordination polymers 1 and 2 was ascertained by comparing the experimental PXRD patterns with the respective simulated powder patterns obtained from the single crystal data. The experimental and simulated PXRD patterns corroborate nicely indicating the phase purity of the bulk products (ESI†S13 and S14).
Crystal and molecular structure of L1 and L2
Ligands L1 (bis(pyridine-3-ylmethyl)terephthalate) and L2 (bis(pyridine-2-ylmethyl)terephthalate) were expediently prepared by slow addition of terephthaloyl chloride into a dichloromethane solution of pyridine-3-methanol or pyridine-2-methanol and triethyl amine. Despite the commercially available starting materials and established esterification routes, ligand L1 was previously unexplored; however, isomeric ligand L2 has been previously reported.13 Colourless shiny single crystals of L1 and L2 suitable for X-ray crystallography were harvested from acetonitrile solution. A summary of the crystal data and selected bond lengths and angles for L1, L2 and their corresponding Ag(I) complexes are given in Table S1 and Table S2.† ORTEP diagrams, along with packing and hydrogen bonding interactions, for L1 and L2 are depicted in Fig. 1 and Fig. 2 respectively.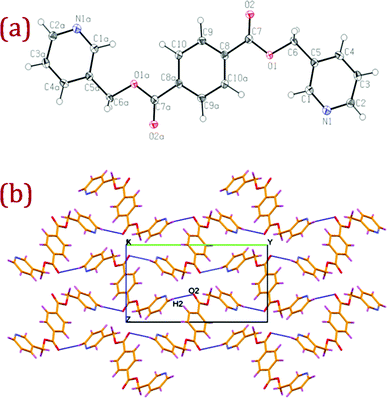 | ||
| Fig. 1 (a) ORTEP diagram of L1 with atom numbering scheme (40% probability factor for the thermal ellipsoids). Additional "a" letters in the atom labels indicate that these atoms are at equivalent position (3 − x, −y, 1 − z); (b) packing diagram of L1 with hydrogen bonding interactions viewed down a-axis showing the hydrogen bonded two dimensional network. Atom labelled as O2 is at equivalent position (3/2 − x, 1/2 + y, 3/2 − z). | ||
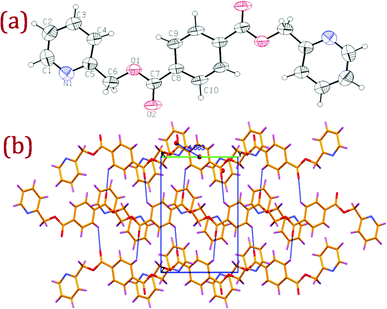 | ||
| Fig. 2 (a) ORTEP diagram of L2 with atom numbering scheme (40% probability factor for the thermal ellipsoids); (b) packing diagram of L2 with hydrogen bonding interactions viewed down a-axis showing the hydrogen bonded two dimensional network. | ||
L1 and L2 crystallized in monoclinic space groups P21/n and P21/c respectively. In both the ligands, the terminal pyridyl rings show anti conformation and are mutually related by center of inversion at the midpoint of benzene ring. Both the ligands adopted an extended conformation and distances between the pyridine nitrogen atoms are 13.70 and 14.30 Å in L1 and L2 respectively. Packing of L1 viewed down the a-axis clearly shows that the adjacent molecules are arranged almost perpendicular to each other and oriented diagonal to the bc-plane to make effective C–H⋯O hydrogen bonding interactions generating a two dimensional hydrogen bonded network as depicted in Fig. 1(b).
Thus, the symmetry related terminal pyridyl hydrogen H2 is making a closer approach to the ester oxygen O2 within the perpendicular ligand moieties making good intermolecular C–H⋯O interaction (C(2)–H(2)⋯O(2): H(2)⋯O(2) = 2.48(3) Å, C(2)⋯O(2) = 3.362(3) Å, ∠ C(2)–H(2)⋯O(2) = 157(2)° with symmetry code:(3/2 − x, 1/2 + y, 3/2 − z)) generating a hydrogen bonded two dimension sheet-like architecture.
The L2 packing diagram viewed down the a-axis revealed that the molecules are aligned along the b-axis with the symmetrically disposed terminal pyridyl rings making an offset π⋯π stacking with the central phenyl ring of adjacent molecules with a centroid⋯centroid distance 4.08 Å with an inter planar distance between the stacked rings of 3.54 Å. Adjacent layers of these stacked molecules are involved in weak intermolecular C–H⋯O interactions along the c-axis between the ester oxygen O2 and phenyl hydrogen H9 of the centrosymmetric molecule with the following hydrogen bonding parameters (C(9)–H(9)⋯O(2): H(9)⋯O(2) = 2.56(3) Å; C(9)⋯O(2) = 3.191(4) Å; ∠ C(9)–H(9)⋯O(2) = 126(2)°; symmetry code: x, 1/2 − y, 1/2 + z)) generating a two dimensional hydrogen bonded network as depicted in Fig. 2(b).
Crystal and molecular structure of {[Ag(L1)(μ2–CH3CN)]·[Ag(L1)(CF3SO3)]·CF3SO3}n (1)
The reaction of AgCF3SO3 with bis(pyridine-3-ylmethyl)terephthalate (L1) in an equimolar ratio leads to the formation of the silver(I) assembly. The crystal structure of complex 1 revealed the presence of two different 1-D extended arrangements: a neutral single chain of [Ag(L1)(CF3SO3)]n (denoted as chain A) and a cationic double-chain [Ag(L1)(μ2–CH3CN)]n (denoted as chain B) as depicted in Fig. 3. In the neutral single chain A, ligand L1 adopts a trans conformation and connects with two Ag(I) centres through the terminal pyridyl nitrogens forming an infinite zigzag chain along the b-axis as depicted in Fig. 3(a). The Ag(I) center in chain A is three coordinated; out of three, two come from pyridyl N atoms [(Ag(1)–N(1) = 2.168(2) Å; (Ag(1)–N(2) = 2.168(2) Å] and the third from triflate oxygen O14 [(Ag(1)–O(14)=2.620(2) Å] rendering T-shaped geometry with the Ag⋯Ag distance connected by L1 being 16.22 Å. The three angles around the metal center Ag(1) are N(2)–Ag(1)–N(1) = 161.92(9)°, N2–Ag1–O14 = 98.83(9)° and N1–Ag1–O14 = 89.54(8)° and the Ag(1) deviate from their corresponding coordination plane by 0.282 Å. Adjacent neutral strands A are related by an inversion centre with a weak offset stacking interaction between the aromatic rings oriented along the b-axis.![ORTEP diagram of neutral single chain [Ag(L1)(CF3SO3)]n (chain A) and cationic double-chain [Ag(L1)(μ2–CH3CN)]n (chain B) present in complex 1 with atom numbering scheme (40% probability factor for the thermal ellipsoids). Atoms labelled as prime, i.e., Ag1′, Ag2′ and N3′ are at equivalent position (−1 + x, y, 1 + z), Atom labelled as N5′′ is at equivalent position (−x, −y, −z).](/image/article/2012/RA/c2ra20642k/c2ra20642k-f3.gif) | ||
| Fig. 3 ORTEP diagram of neutral single chain [Ag(L1)(CF3SO3)]n (chain A) and cationic double-chain [Ag(L1)(μ2–CH3CN)]n (chain B) present in complex 1 with atom numbering scheme (40% probability factor for the thermal ellipsoids). Atoms labelled as prime, i.e., Ag1′, Ag2′ and N3′ are at equivalent position (−1 + x, y, 1 + z), Atom labelled as N5′′ is at equivalent position (−x, −y, −z). | ||
Both terminal heterocyclic pyridyl rings (C1 to C5–N1 and C16 to C20–N2) of the coordinated ligand L1 are not co-planar and form dihedral angles of 46.29° and 26.84° with respect to the central phenyl ring (C8–C13) to make efficient coordination with the metal centre. In the second co-existing building block, the cationic double-chain [Ag(L1)(μ2–CH3CN)]n, silver(I) (chain B) is co-ordinated to nitrogen of L1 from either end generating a one dimensional single strand with trans orientation of the terminal pyridyl rings. Two such adjacent single strands are bridged through the metal centre via a μ2-coordination mode of two acetonitrile molecules sandwiched between the single strands in proximity to the metal ion forming the cationic double-chain [Ag(L1)(μ2–CH3CN)]n assembly of the extended network as depicted in Fig. 3. The metallic ion occupies a general position, leading to an almost linear N3–Ag2–N4 (∠ N3–Ag2–N4 = 174.20(8)°) co-ordination geometry within the single stranded chain and the whole double-chain strand possesses a centre of inversion at the middle of Ag⋯Ag metal ions of the neighbouring mono strands bridged via μ2-acetonitrile coordination. Ag⋯N distances involving the pyridyl linker N3 and N4 are 2.186(2) and 2.188(2) Å while N5 that of the acetonitrile linking with Ag(1) of the adjacent strands within the double chain assembly is weaker and comparatively longer in nature (Ag(2)–N(5) = 2. 716(2) Å; Ag(2a)–N(5) = 2.717(3) Å). The nearest non-bonding Ag2⋯Ag2 distance connected by L1 is 16.22 Å, which is in the same range as the single chain and the closest Ag2⋯Ag2 distance within the center of the symmetric double chain assembly bridged through μ2–N(5) of the acetonitrile is 3.56 Å, which is slightly more (by 0.12 Å) than the summed van der Waals radii of two silver atoms (3.44 Å), indicating the existence of weak Ag⋯Ag interactions within the double stranded chain.14,15 The terminal pyridyl rings (C21to C25–N3) and (C36 to C40–N4) of L1 are rotated by 38.39° and 30.82° with respect to the central phenyl ring (C28 to C32) to make effective coordination with the Ag(2) cation.
In an attempt to understand the arrangement of the chains A and B in the crystal lattice, packing and hydrogen bonding interactions were investigated in detail. The orientation of the single neutral chain A and double chain B along the a-axis is depicted Fig. 4. Within the centrosymmetric double chain, L1 of the adjacent strands show slightly offset intra chain π⋯π stacking interaction with centroid⋯centroid distances between the terminal pyridyl rings of 3.91 Å and the central aryl rings of 3.89 Å respectively. The closer approach of the aromatic ring for better π⋯π stacking is mediated by the μ2-coordination of the acetonitrile molecule bridging the neighbouring strands. Two more neutral strands of chain A are assembled on top of the cationic double chain B, which are further arranged periodically resulting in an AAB⋯AAB sequence of coordination array along the ab plane as depicted in Fig. 4. Two adjacent chains A and B are associated by the inter chain offset π⋯π stacking interactions between the aromatic rings of L1 in which the centroid⋯centroid distances between the terminal pyridyl rings are 3.78 Å and 3.81 Å respectively and that between stacked central phenyl rings is 3.95 Å.16 Interestingly, no π⋯π stacking interaction is observed in the supramolecular arrangement of L1 in the crystallographic investigation. Notably, the neighbouring inter chain Ag1⋯Ag2 separation is 3.364(2) Å (between chain A and chain B), which is shorter than that of van der Waals Ag⋯Ag interaction (3.44 Å) and longer than that of metallic silver (2.89 Å), indicating the presence of inter-chain argentophilic interactions (Fig. 4).6,15
![View of the different types of 1D chains A (neutral single strands) and B (cationic double strand) along [100] in complex 1, showing the coordination environments of Ag1 and Ag2 (cyan spheres) in the chain as well as intra/inter chain π⋯π stacking (blue dashed lines) and Ag⋯Ag (blue dashed lines) interactions.](/image/article/2012/RA/c2ra20642k/c2ra20642k-f4.gif) | ||
| Fig. 4 View of the different types of 1D chains A (neutral single strands) and B (cationic double strand) along [100] in complex 1, showing the coordination environments of Ag1 and Ag2 (cyan spheres) in the chain as well as intra/inter chain π⋯π stacking (blue dashed lines) and Ag⋯Ag (blue dashed lines) interactions. | ||
Within the aforementioned AAB motifs although Ag1⋯Ag1 separation of the nearby single stranded A chains is 5.12 Å, inter-chain stacking of L1 is observed. Thus, the centroid⋯centroid distance of the terminal pyridyl which is making weak off set π⋯π stacking is 4.23 Å whereas the central phenyl rings are engaged in good stacking with a centroid⋯centroid distance of 3.68 Å.16 Packing down the a-axis for 1 revealed C–H⋯O interactions: ester oxygen of the ligand with the aromatic hydrogens/methylene hydrogens; sulfonate oxygens from the coordinated and lattice triflate anions with the aromatic hydrogens and the methyl hydrogens of the acetonitrile molecules respectively (Table S2†). Thus, the coordinated sulfonate oxygen O13 and lattice sulfonate oxygen O9 from triflate are involved in intramolecular C–H⋯O interaction with the phenyl hydrogens H1 and H21 of the coordinated terminal pyridyl rings of L1 coordinated to Ag1 and Ag2 with D⋯A distances of 3.183(3) and 3.160(4) Å respectively. Phenyl and methylene hydrogens (H3 and H6A) of the stacked single and double strand chains down the b-axis (in AAB sequence) are involved in bifurcated intermolecular C–H⋯O interactions with the carbonyl oxygen O3 and the second carbonyl oxygen O2 of L1, making C–H⋯O contact with the methylene hydrogen H15A of the single strand ligand moiety of the neighbouring AAB sequence chains with D⋯A distances ranging from 3.215(3) to 3.348(3) Å. Uncoordinated triflate oxygens O9, O10 and O11 are involved in intermolecular C–H⋯O H–bonds with pyridyl hydrogen H21 of the double stranded chain B, pyridyl hydrogen of the single strand chain A and the phenyl hydrogen H30 of the double stand chain with D⋯A distances ranging from 3.160(4) to 3.272(5) Å in linking the π stacked sequential AAB strands. Further, O11 is also involved in an intramolecular C–H⋯O contact with the μ2-coordinated acetonitrile hydrogen H43A (D⋯A = 3.320(10) Å). O12 is making a bifurcated intramolecular C–H⋯O contact with H20 of chain A, H33 phenyl hydrogen of chain B while O13 is involved with pyridyl hydrogen H38 of L1 (in chain B) with D⋯A distances ranging from 3.237(3) to 3.485(4) Å. Details of all pertinent hydrogen bonding interaction with symmetry code is given in Table S2 of the ESI.† π⋯π Stacking between the aromatic moiety of L1 within the AAB sequential strands coupled with C–H⋯O interaction involving the triflate/carboxyl oxygen of L1 as acceptor concomitantly stabilizes the supramolecular three dimensional network in the crystal lattice.
Crystal and molecular structure of {[Ag(L2)]n·CF3SO3·H2O} (2)
When L1 is replaced by isomeric ligand L2, we obtain a 1D cationic zigzag coordination polymer, which crystallizes in the orthorhombic space group Pbcn. Each asymmetric unit is composed of Ag(I) ion, half a molecule of L2 with the center of symmetry passing through the central phenyl ring, triflate anion and water molecule as solvent of crystallization. Analysis of the local symmetry of the metal atoms, ligand L2, anion and lattice water shows that all of them are located in special positions. In detail, a C2 axis passes through the triflate anion along S1 and C11 atoms (and bisecting the F1 and O10) and also bisects water oxygen O4. The Ag(I) cation is also located at a special position. Explicitly, space-group imposed symmetry offers Ag1, S1, C11 atoms to lie on a twofold axis, water oxygen O8 on an adjacent twofold axis, and an inversion centre at the middle of the central aromatic ring in L2. Nevertheless, the crystal structure of the same compound in a different polymorphic from (space group P21/n) reported by Son et al. is worth mentioning here.13 Crystallographic analysis, including the packing of the new polymorphic form, is examined in detail to gain some insight towards the room temperature fluorescence displayed by the Ag(I) complex. An ORTEP diagram of the zigzag one dimensional cationic coordination polymer with atom numbering scheme is given in the ESI† as S18. [Ag(L2)]nCF3SO3·H2O (2) consists of a zigzag polymeric cationic 1D chain and CF3SO3 anions along with one water molecule as solvent of crystallization in which each Ag(I) center is linked by symmetrically disposed N donor ligand [Ag–N = 2.129(3) Å] (Fig. 5 (a)). In 2, ligand L2 is almost planar and adopts an extended conformation as observed in the crystal structure of the ligand moiety. The N–Ag–N [175.55(14)°] angle confirms a linear environment around the Ag(I) center with the nearest non-bonding Ag⋯Ag distance connected by L2 being 16.38 Å.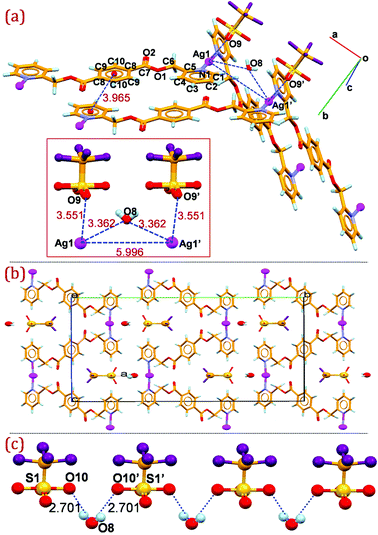 | ||
| Fig. 5 (a) Mercury diagram depicting the non-bonding molecular interaction between the adjacent strands in 2 (intermolecular distances are presented in the inset); (b) packing diagram of 2 viewed down the a-axis showing the corrugated arrangement of the cationic strands and the orientation of the triflate anions and water molecules within the clefts; (c) O–H⋯O hydrogen bonding interactions between the lattice water molecules and triflate anions in 2. Atoms labelled as prime are at equivalent position (−x, y, 3/2 − z). | ||
As depicted in Fig. 5 (a) non-bonding molecular interactions do exist between the adjacent strands wherein the lattice water molecule is oriented between the adjacent strands with closer proximity towards the silver cation on either side making short contact with the Ag(I)⋯O8 distance 3.36 Å almost bridging the neighbouring stacks. It is also observed that O9 from the triflate anion is also in the vicinity of Ag(I) at a distance Ag1⋯O9 = 3.551(4) Å, making weak contact. The neighbouring strands are oriented to make effective π⋯π stacking between the symmetrically disposed pyridyl ring of one strand with the central phenyl ring of the adjoining strand, with a π⋯π stacking distance between the centroid of the rings of 3.96 Å and an interplanar distance between the stacked rings of 3.65 Å, as represented in Fig. 5 (a). Notably, the neighbouring Ag1⋯Ag1 separation between the adjacent zigzag chain is 5.996(4) Å, indicative of the absence of argentophilic interactions. The packing diagram viewed down the a-axis is depicted in Fig. 5 (b).
Zigzag silver strands coordinated through the N-donor ligands are stacked down the a-axis generating clefts which the triflate anions and water molecule occupy. Symmetrically disposed water hydrogens are involved in O–H⋯O interaction of the sulfonate oxygen of the triflate anion placed on two fold axis to generate hydrogen bonded zigzag chain down the a-axis (Fig. 5 (c)). In addition to the weak non-bonding interaction of the water molecule with the silver cation between the strands, the oxygen atom of the water molecule O8 act as an acceptor and is involved in C–H⋯O interaction with the methylene carbon H(6A) (C(6)–H(6A)⋯O(8): H(6A)⋯O(8)=2.51 Å; C(6)⋯O(8)=3.413(8) Å; ∠ C(6)–H(6A)⋯O(8)=154°) in bridging the neighbouring coordination polymer. H6B of the methylene group is also making intermolecular C–H⋯O hydrogen bond with the carbonyl oxygen O2 (C(6)–H(6B)⋯O(2): H(6B)⋯O(2) = 2.47 Å; C(6)⋯O(2) = 3.317(4) Å; ∠ C(6)–H(6B)⋯O(2) = 146°) of the ligand moiety, generating a supramolecular two dimensional hydrogen bonded network in the bc-plane.
Photoluminescence study
The luminescent behaviour of ligands L1 and L2 as well as complexes 1 and 2 were studied at room temperature in the solution and solid states (Fig. 6). Acetonitrile solutions of 1, 2, L1 and L2 show absorbance maxima at ca. 242 nm at room temperature (ESI†S15–S17). When excited to 242 nm in solution an emission maximum at 319 nm was observed for both complexes 1 and 2, which can be attributed to the intraligand transmissions. The absorption and emission behaviour of 1 and 2 in solution is quite comparable with their respective ligands L1 and L2, suggesting ligand centred π*–π and/or π*–p transmissions.17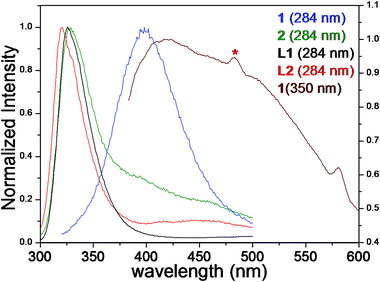 | ||
| Fig. 6 Normalized emission (λex 284) profiles of L1, L2, 1 and 2 recorded in the solid state using crystalline powder samples prepared by gentle crushing of crystals. Additional emission spectrum (λex 350) for 1 showing vibronic band at ca. 482 nm is indicative of argentophilic interactions. | ||
The absorption spectra of compounds 1, 2, L1 and L2 in the solid state show absorption peaks at ca. 284 and 300 nm. On excitation at 284 nm in the solid state complex 2, L1 and L2 exhibited phosphorescent emissions at 321, 326 and 328 nm respectively. These emissions are comparable with those obtained in acetonitrile solutions and can be ascribed to ligand centred π*–π and/or π*–p transmissions.
Upon excitation of complex 1 at 284 nm in the solid state blue emission with a peak maximum at 398 nm was observed. This emission band is considerably red-shifted (over 75 nm) compared with either free ligand or complex 2. It is noteworthy that the complex 1 in acetonitrile solution does not show such a substantial red shift. The emission spectra of complex 1 persists even after heating the sample up to 150 °C which rules out any possibility of solvatochromic or vapochromic emission.18 Obviously the red-shifted emission of complex 1 can be attributed to the collective contribution of solid state structural features such as framework robustness, π⋯π stacking and argentophilic interactions.7,19 It is evident from the crystallographic studies that complex 1 contains coordinated silver(I) centres in close proximity (between the chains A and B and within the double stranded B) i.e., Ag⋯Ag separations (3.364(2) Å in 1) are either within the range of argentophilic interactions or slightly above the upper limit.
Consequently, the observed emission band at ca. 398 nm for 1 is ascribed to the concomitant effect of argentophilic cluster centred emission,4j,20 the metal to ligand charge transfer (MLCT)21 and counter anion to ligand charge transfer (XLCT) transmissions.22 In order to further ascertain the connection between crystallographically established short Ag⋯Ag separation and luminescent behaviour another excitation wavelength (350 nm) was obtained from excitation spectra (emission 400 nm). The emission profile showed a maximum wavelength (λem max) at 418 nm, accompanied by a weaker vibronic band at ca. 482 nm (marked by an asterisk in Fig. 6). The vibronic band at 482 nm was attributed to the presence of argentophilic interactions in the silver coordination polymer 1. In this regard, it is worth mentioning that similar emission behaviour has been reported in the literature for silver and gold complexes.23 Markedly, a direct correlation between the emission wavelength and crystallographic Ag⋯Ag distance in silver complexes is difficult as such interactions are not yet fully understood. Ag(I) compounds usually exhibit poor photoluminescence even at low temperature due to the spin–orbital coupling of Ag(I). As complex 1 emits blue photoluminescence in the solid state at ambient temperature it may be a good candidate for photoluminescent materials.
Conclusion
In summary, crystal structures of two isomeric ligands L1, L2 and their Ag(I) coordination compounds 1, 2 were synthesized and structurally characterized. Crystal structure analysis shows that 1 is composed of two distinct coordination polymers, neutral T-shaped single chain and a μ2-acetonitrile bridged symmetric double chain with Ag⋯Ag separation (3.364(2) Å) falling within the accepted range for argentophilic interaction. Complex 2 crystallized in a different polymorphic form and is comprised of 1D zigzag chains. Both Ag complexes showed room temperature photoluminescent property, particularly complex 1 exhibits strong blue emission which can be attributed to the stacking interactions of the ligand moiety and argentophilic interaction between the single and double chains. Mixed ligand Ag(I) complexes could be well tuned by ligand modifications and efforts are in progress to design new Ag(I) complexes with versatile structural topology and photoluminescence properties with the same isomeric N-donor ligands in conjunction with carboxylate linkers.Acknowledgements
Authors gratefully acknowledge the CSIR, India (Grant No. NWP-0010) for financial support, Dr Vinod P. Boricha for NMR data, Dr Pragnya A. Bhatt for XRPD data, Mrs. Sheetal N. Patel for TGA data, Mr. Vinod Kumar Agrawal for IR data, Mr. Viral Vakani for microanalysis and Dr P. Paul for all-round analytical support. KKB acknowledges CSIR (India) for SRF.References
- (a) J. A. Ramsden, W. Weng, A. M. Arif and J. A. Gladysz, J. Am. Chem. Soc., 1992, 114, 5890–5891 CrossRef CAS; (b) M. Fujita, Y. J. Kwon, S. Washizu and K. J. Ogura, J. Am. Chem. Soc., 1994, 116, 1151–1152 CrossRef CAS; (c) N. Le Narvor, L. Toupet and C. Lapinte, J. Am. Chem. Soc., 1995, 117, 7129–7138 CrossRef CAS; (d) T. Bartik, W. Weng, J. A. Ramsden, S. Szafert, S. B. Falloon, A. M. Arif and J. A. Gladysz, J. Am. Chem. Soc., 1998, 120, 11071–11081 CrossRef CAS; (e) K. Biradha, C. Seward and M. J. Zaworotko, Angew. Chem., 1999, 111, 584–587 CrossRef; (f) A. Tanatani, M. J. Moi and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 1792–1793 CrossRef CAS; (g) O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (h) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS; (i) B. F. Hoskins, R. Robson and D. A. Slizys, J. Am. Chem. Soc., 1997, 119, 2952–2953 CrossRef CAS; (j) N. R. Brooks, A. J. Blake, N. R. Champness, J. W. Cunningham, P. Hubberstey and M. Schröder, Cryst. Growth Des., 2001, 1, 395–399 CrossRef CAS; (k) B. Schmaltz, A. Jouaiti, M. W. Hosseini and A. D. Cian, Chem. Commun., 2001, 1242–1243 RSC; (l) A. L. Pickering, G. Seeber, D.-L. Long and L. Cronin, Chem. Commun., 2004, 136–137 RSC; (m) J. Bourlier, M. W. Hosseini, J.-M. Planeix and N. Kyritsakas, New J. Chem., 2007, 31, 25–32 RSC; (n) S. Ma and H. –C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC; (o) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS; (p) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688 CrossRef CAS.
- (a) G. K. Kole, G. Kheng Tan and J. J. Vittal, Cryst. Growth Des., 2012, 12, 326–332 CrossRef CAS; (b) C.-Q. Wan and T. C. W. Mak, Cryst. Growth Des., 2011, 11, 832–842 CrossRef CAS; (c) C.-Q. Wan and T. C. W. Mak, New J. Chem., 2011, 35, 319–327 RSC; (d) B. Li, S.-Q. Zang, C. Ji, C.-X. Du, H.-W. Hou and T. C. W. Mak, Dalton Trans., 2011, 40, 788–792 RSC; (e) G. K. Patra and I. Goldberg, Cryst. Growth Des., 2003, 3, 321–329 CrossRef CAS; (f) A. Bellusci, M. Ghedini, L. Giorgini, F. Gozzo, E. I. Szerb, A. Crispini and D. Pucci, Dalton Trans., 2009, 7381–7389 RSC; (g) A. K. Mishra, R. K. Prajapati and S. Verma, Dalton Trans., 2010, 39, 10034–10037 RSC; (h) S. Wang, H. Zang, C. Sun, G. Xu, X. Wang, K. Shao, Y. Lan and Z. Su, CrystEngComm, 2010, 12, 3458–3462 RSC; (i) T. H. Noh, Y. J. Choi, Y. K. Ryu, Y.-A. Lee and O.-S. Jung, CrystEngComm, 2009, 11, 2371–2374 RSC; (j) M. Du, C.-P. Li and J.-H. Guo, CrystEngComm, 2009, 11, 1536–1540 RSC; (k) C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
- (a) M. Munakata, L. P. Wu and G. L. Ning, Coord. Chem. Rev., 2000, 198, 171 CrossRef CAS; (b) A.G. Young and L. R. Hanton, Coord. Chem. Rev., 2008, 252, 1346–1386 CrossRef CAS; (c) P. J. Steel, Acc. Chem. Res., 2005, 38, 243 CrossRef CAS; (d) J. L. Sagué and K. M. Fromm, Cryst. Growth Des., 2006, 6, 1566 CrossRef; (e) A. Erxleben, Inorg. Chim. Acta, 2003, 348, 107–114 CrossRef CAS; (f) G.-P. Yang, J.-H. Zhou, Y.-Y. Wang, P. Liu, C.-C. Shi, A.-Y. Fu and Q.-Z. Shi, CrystEngComm, 2011, 13, 33–35 RSC; (g) G. Yuan, C. Zhu, Y. Liu, W. Xuan and Y. Cui, J. Am. Chem. Soc., 2009, 131, 10452–10460 CrossRef CAS; (h) C. Desmarets, I. Azcarate, G. Gontard and H. Amouri, Eur. J. Inorg. Chem., 2011, 4558–4563 CrossRef CAS; (i) A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schröder, Coord. Chem. Rev., 2001, 222, 155 CrossRef CAS; (j) D. Sun, Z.-H. Wei, D.-F. Wang, N. Zhang, R.-B. Huang and L.-S. Zheng, Cryst. Growth Des., 2011, 11, 1427–1430 CrossRef CAS.
- (a) D. Pucci, A. Crispini, M. Ghedini, E. I. Szerb and M. L. Deda, Dalton Trans., 2011, 40, 4614–4622 RSC; (b) I. Stoll, R. Brockhinke, A. Brockhinke, M. Böttcher, T. Koop, H.-G. Stammler, B. Neumann, A. Niemeyer, A. Hütten and J. Mattay, Chem. Mater., 2010, 22, 4749–4755 CrossRef CAS; (c) H. Yang, Y.-N. Lao, J.-M. Chen, H.-X. Wu and S.-P. Yang, Eur. J. Inorg. Chem., 2009, 2817–2824 CrossRef CAS; (d) S.-M. Fang, M. Hu, Q. Zhang, M. Du and C.-S. Liu, Dalton Trans., 2011, 40, 4527–4541 RSC; (e) H.-J. Hao, D. Sun, Y.-H. Li, F.-J. Liu, R.-B. Huang and L.-S. Zheng, Cryst. Growth Des., 2011, 11, 3564–3578 CrossRef CAS; (f) Y. Ling, Z.-X. Chen, Y.-M. Zhou, L.-H. Weng and D.-Y. Zhao, CrystEngComm, 2011, 13, 1504–1508 RSC.
- (a) C. J. Shorrock, B.-Y. Xue, P. B. Kim, R. J. Batchelor, B. O. Patrick and D. B. Leznoff, Inorg. Chem., 2002, 41, 6743–6753 CrossRef CAS; (b) W. G. Lu, J. Z. Gu, L. Jiang, C. Y. Jiang, C. Y. Su and T. B. Lu, Chin. J. Inorg. Chem., 2006, 22, 1977–1981 CAS; (c) Z. X. Lian, J. Cai, C. H. Chen and H. B. Luo, Cryst. Eng. Commun., 2007, 9, 319–327 CAS; (d) B. Liu, W. Chen and S. Jin, Organometallics, 2007, 26, 3660–3667 CrossRef CAS; (e) L. Valencia, R. Bastida, A. Macías, M. Vicente and P. Pérez-Lourido, New J. Chem., 2005, 29, 424–426 RSC; (f) A. Serpe, F. Artizzu, L. Marchiò, M. L. Mercuri, L. Pilia and P. Deplano, Cryst. Growth Des., 2011, 11, 1278–1286 CrossRef CAS; (g) J. Moussa, K. Boubekeur and H. Amouri, Eur. J. Inorg. Chem., 2005, 3808–3810 CrossRef CAS; (h) L. Dai, W. You, E. Wang, S. Wu, Z. Su, Q. Du, Y. Zhao and Y. Fang, Cryst. Growth Des., 2009, 9, 2110–2116 CrossRef CAS; (i) Y.-B. Xie, Q. Gao, C.-Y. Zhang and J.-H. Sun, J. Solid State Chem., 2009, 182, 1761–1766 CrossRef CAS.
- S. Q. Liu, T. K.-Sowa, H. Konaka, Y. Suenaga, M. Maekawa, T. Mizutani, G. L. Ning and M. Munakata, Inorg. Chem., 2005, 44, 1031–1036 CrossRef CAS.
- (a) A. C. Kathalikkattil, K. K. Bisht, N. Aliaga-Alcalde and E. Suresh, Cryst. Growth Des., 2011, 11, 1631–1641 CrossRef CAS; (b) A. C. Kathalikkattil, K. K. Bisht, P.S. Subramanian and E. Suresh, Polyhedron, 2010, 29, 1801–1809 CrossRef CAS; (c) A. C. Kathalikkattil, P.S. Subramanian and E. Suresh, Inorg. Chim. Acta, 2011, 365, 363–370 CrossRef CAS; (d) A. C. Kathalikkattil, S. Damodaran, K. K. Bisht and E. Suresh, J. Mol. Struct., 2011, 985, 361–370 CrossRef CAS; (e) K. K. Bisht, A. C. Kathalikkattil and E. Suresh, Cryst. Growth Des., 2012, 12, 556–561 CrossRef CAS; (f) K. K. Bisht, A. C. Kathalikkattil and E. Suresh, J. Mol. Struct., 2012, 1013, 102–110 CrossRef CAS.
- G. M. Sheldrick, SAINT 5.1 ed., Siemens Industrial Automation Inc., Madison, WI, 1995 Search PubMed.
- SADABS, Empirical Absorption Correction Program, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXTL Reference Manual: Version 5.1, Bruker AXS, Madison,WI, 1997.
- G. M. Sheldrick, SHELXL-97: Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- M. K. Kalra, H. Zhang and D. Y. Son, Inorg. Chem. Comm., 2004, 7, 1019–1022 CrossRef CAS.
- A. J. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) V. W. W. Yam, P. K. Y. Yeung and K. K. Cheung, Angew. Chem., Int. Ed. Engl., 1996, 35, 739 CrossRef CAS; (b) D. Venkataraman, Y. Du, S. R. Wilson, K. A. Hirsch, P. Zhang and J. S. Moore, J. Chem. Educ., 1997, 74, 915 CrossRef CAS; (c) M. L. Tong, X. M. Chen and B. H. Ye, Inorg. Chem., 1998, 37, 5278 CrossRef CAS; (d) M. L. Tong, S. L. Zheng and X. M. Chen, Chem. Commun., 1999, 561 RSC; (e) I. Yoon, Y. H. Lee, J. H. Jung, K. M. Park, J. Kim and S. S. Lee, Inorg. Chem. Commun., 2002, 5, 820 CrossRef CAS; (f) C.-P. Li, J. Chen, Q. Yu and M. Du, Cryst. Growth Des., 2010, 10, 1623–1632 CrossRef CAS; (g) X. Liu, G.-C. Guo, M.-L. Fu, X.-H. Liu, M.-S. Wang and J.-S. Huang, Inorg. Chem., 2006, 45, 3679 CrossRef CAS; (h) M. L. Tong, X. M. Chen, B. H. Ye and L. N. Ji, Angew. Chem., Int. Ed., 1999, 38, 2237 CrossRef CAS; (i) C.-M. Che, M.-C. Tse, M. C. W. Chan, K.-K. Cheung, D. L. Phillips and K.-H. Leung, J. Am. Chem. Soc., 2000, 122, 2464 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC and references therein.
- (a) Y. B. Dong, H. Y. Wang, J. P. Ma, D. Z. Shen and R. Q. Huang, Inorg. Chem., 2005, 44, 4679 CrossRef CAS; (b) C. D. Wu, H. L. Ngo and W. B. Lin, Chem. Commun., 2004, 1588 RSC; (c) J.-L. Du, T.-L. Hu, S.-M. Zhang, Y.-F. Zeng and X.-H. Bu, CrystEngComm, 2008, 10, 1866–1874 RSC; (d) H. Wu, X. W. Dong, H. Y. Liu, J. F. Ma, S. L. Li, J. Yang, Y. Y. Liu and Z. M. Su, Dalton Trans., 2008, 5331 RSC.
- (a) A. Bergen, C. Bohne, D. Fuentealba, H. Ihmels, T. C. S. Pace, M. Waidelich, C. Yihwa and J. W. Bats, Photochem. Photobiol. Sci., 2012, 11, 752 RSC; (b) E. J. Rivera, C. Barbosa, R. Torres, L. Grove, S. Taylor, W. B. Connick, A. Clearfield and J. L. Colón, J. Mater. Chem., 2011, 21, 15899–15902 RSC.
- M.-L. Ho, C.-H. Shih, C.-H. Lee and G.-H. Lee, CrystEngComm, 2011, 13, 992–1002 RSC and references therein.
- V. W. W. Yam and K. K. W. Lo, Chem. Soc. Rev., 1999, 28, 323 RSC.
- P.-X. Yin, J. Zhang, Z.-J. Li, Y.-Y. Qin, J.-K. Cheng, L. Zhang, Q.-P. Lin and Y.-G. Yao, Cryst. Growth Des., 2009, 9, 4884–4896 CAS.
- X. C. Huang, S. L. Zheng, J. P. Zhang and X. M. Chen, Eur. J. Inorg. Chem., 2004, 1024–1029 CrossRef CAS.
- (a) L. Ray, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2008, 47, 230–240 CrossRef CAS; (b) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Synthetic route for L1, characterization data for L1, L2, 1 and 2; additional crystallographic figures and tables; crystallographic data (CCDC 868386–868389) for compounds L1, L2, 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20642k/ |
| This journal is © The Royal Society of Chemistry 2012 |