Synthesis and properties of ammonium ionic liquids with cyclohexyl substituent and dissolution of cellulose†
Juliusz
Pernak
*a,
Roksana
Kordala
a,
Bartosz
Markiewicz
a,
Filip
Walkiewicz
a,
Mikołaj
Popławski
a,
Anna
Fabiańska
b,
Stefan
Jankowski
b and
Marek
Łożyński
a
aPoznan University of Technology, pl. M. Sklodowskiej-Curie 2, 60-965 Poznan, Poland. E-mail: juliusz.pernak@put.poznan.pl; Tel: +48 (61) 665 36 82
bInstitute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, ul. Zeromskiego 116, 90-924 Lodz, Poland
First published on 24th July 2012
Abstract
We have synthesized alkyl(cyclohexyl)dimethylammonium bromides, formates and acetates, 2-methoxyacetates, 2-(2-methoxyethoxy)acetates, 2-[2-(2-methoxyethoxy)ethoxy]acetates, and 2-ethoxyacetates and assessed their ability to dissolve cellulose. Their physicochemical properties and microbiological activity were determined. Alkyl(cyclohexyl)dimethylammonium cation dimensions of alkyl groups are essential for cellulose solubility and are generally limited to butyl, hexyl, and octyl as effective substituents. In parallel, the alkyl group determines activity of the entire molecule against microbes. The molecule's biological activity against microbes begins when a length of the alkyl substituent begins to block cellulose solubility. We present the computational model of the solubility mechanism, composed of an ensemble of two cellobiose units and (cyclohexyl)hexyldimethylammonium acetate.
Introduction
In the past few years the chemistry of ionic liquids (ILs) has developed tremendously. Initially they were discussed as a replacement for conventional molecular solvents, yet their physical and chemical properties allowed exploitation of their applications from electrochemistry to biology.1–6 The evaluation of ILs proceeds very quickly from the first generation (ILs with unique tunable physical properties) to the second generation (ILs with targeted chemical properties combined with selected physical properties), to the third generation (ILs with targeted biological properties combined with selected physical and chemical properties).7,8 The newest third generation ILs became enriched by phytopharmaceuticals—herbicidal ionic liquids.9–11In 2002, Rogers' group published a pioneering work, showing that 1-alkyl-3-methylimidazolium chlorides were able to dissolve cellulose.12 This publication opened a new field of cellulose research and many scientists all over the world were trying to dissolve this biopolymer in various kinds of ILs and in different conditions. Several ILs were found to be capable of dissolving cellulose, forming highly viscous solutions.13
The ILs contained the cations including imidazolium, pyridinium, pyrrolidinium, ammonium and piperidinium. Nonetheless, the ILs which were studied and found to dissolve cellulose were still primarily composed of imidazolium or pyridinium-based cations with anions of basic character, such as chloride, carboxylates (formate, acetate), dialkylphosphates or phosphonates.14 The regeneration of cellulose from ILs can be achieved by adding an anti-solvent, such as water or protic organic solvents, such as ethanol, into the solution.15–17 The use of ILs in dissolution of cellulose, lignin and wood has been reviewed recently.13,14,18,19 An excellent critical review on ionic liquid processing of cellulose has just been published.20 According to this review, the role of anion is clear in the process of cellulose dissolution. It is also clear, that the cation must play some role, but opinions about it are divided. Some say that the cation has no interactions with the cellulose unit, some say that strong hydrogen bonding between them exists.
Mechanistic studies were conducted on imidazolium and pyridinium cations, because they are most often studied in the dissolution of cellulose. Almost no attention has been paid to other cations.20 It was attempted to define the role of alkyl(cyclohexyl)dimethylammonium cation in the dissolution of cellulose. It was expected that the alkyl group will determine the properties of alkyl(cyclohexyl)dimethylammonium ILs.
Experimental section
Materials
(Cyclohexyl)dimethylamine, bromoalkanes, formic acid, acetic acid, 2-methoxyacetic acid, 2-(2-methoxyethoxy)acetic acid, 2-[2-(2-methoxyethoxy)ethoxy]acetic acid, 2-ethoxyacetic acid, potassium hydroxide, and microcrystalline cellulose (CAS: 9004-34-6) as well as all solvents were purchased from commercial suppliers (Sigma-Aldrich, Fluka) and used without further purification.General
1H NMR spectra were recorded on a Mercury Gemini 300 spectrometer operating at 300 MHz with TMS as the internal standard. 13C NMR spectra were obtained with the same instrument at 75 MHz. CHN elemental analyses were performed at the Adam Mickiewicz University, Poznan (Poland).1H and 13C NMR spectra of alkyl(cyclohexyl)dimethylammonium bromides (1–3), (cyclohexyl)hexyldimethylammonium acetate (15) and its solution with cellulose were recorded at 27 °C in DMSO-d6 (99.8% D, Armar) on a Bruker Avance II Plus spectrometer at 700.21 and 176.09 MHz, respectively. 1H and 13C chemical shifts were calibrated on solvent signals at 2.49 and 39.7 ppm, respectively, or to the external standard CD3NO2 (62.8 ppm) placed in a coaxially placed capillary. 14N-NMR spectra for alkyl(cyclohexyl)dimethylammonium bromides (1–3) and (cyclohexyl)hexyldimethylammonium acetate (15) were recorded at 50.58 MHz. 14N chemical shifts were referred to neat nitromethane with negative signals upfield against the reference. The signal assignments were based on analysis of 1H and 13C 1D NMR, 1H–1H COSY and ROESY, 1H–13C HSQC and HMBC spectra. For ROESY spectra a mixing time of 300 ms was applied.
The water content was determined by using an Aquastar volumetric Karl Fischer titration with Composite 5 solution as the titrant and anhydrous methanol as a solvent. Melting points were determined by visual observation via hot-plate apparatus.
Density was determined using an Automatic Density Meter DDM2911 with a mechanical oscillator method. The density of the samples, about 2.0 mL, was measured with respect to temperature controlled conditions via Peltier, at 25 °C. The apparatus used was calibrated using deionized water as the reference substance. After each series of measurements, the densimeter was washed by two kinds of solvents and dried.
Viscosity was determined using a rheometer (Rheotec RC30-CPS) with cone-shaped geometry (C50-2). The viscosity of the samples, about 1.5 mL was measured with respect to temperature, from 20 to 100 °C. The uncertainty of the viscosity measurement was estimated to be less than 10−4 Pa s. Refractive index was determined using Automatic Refractometer J357 with electronic temperature control.
Preparation of alkyl(cyclohexyl)dimethylammonium bromides (1–9)
Cyclohexyldimethylamine (0.1 mol) was dissolved in acetonitrile in a round bottom flask. The appropriate bromoalkane was added in 5% extension and the mixture was stirred at room temperature for 24 h. By evaporation of the reaction mixture, at a temperature of 40 °C, the crude product was obtained. Ethyl acetate was added, and the pure product was isolated by filtering and dried over night at 40 °C under reduced pressure.Descriptions of NMR spectra and elemental analysis for bromides (1, 2, 4–9) can be found in the Supplementary Information.†
Synthesis of alkyl(cyclohexyl)dimethylammonium salts (10–30)
Alkyl(cyclohexyl)dimethylammonium bromide (0.05 mol) was dissolved in methanol and a stoichiometric amount of saturated methanolic solution of KOH was added. The solutions were stirred at room temperature for 5 min, after which the partially precipitated KBr was filtrated. Then a stoichiometric amount of an appropriate alkylcarboxylic acid was added. The solutions were stirred again at room temperature for 15 min and after evaporation of the solvent at a temperature of 40 °C, the product was dissolved in 20 mL of anhydrous acetone. The inorganic salt was separated by filtering and acetone was evaporated to give the product, which was finally dried under reduced pressure at 80 °C for 24 h.Descriptions of NMR spectra and elemental analysis for the synthesized salts 10–14 and 16–30 can be found in the Supplementary Information.†
Thermal analysis
Thermal transitions of prepared salts were determined by DSC, with a Mettler Toledo Stare TGA/DSC1 (Leicester, UK) unit, under nitrogen. Samples between 5 and 15 mg were placed in aluminium pans and heated from 25 to 120 °C at a heating rate of 10 °C min−1 and cooled with an intracooler at a cooling rate of 10 °C min−1 to −100 °C min−1. Thermogravimetric analysis was performed using a Mettler Toledo Stare TGA/DSC1 unit (Leicester, UK) under nitrogen. Samples between 2 and 10 mg were placed in aluminium pans and heated from 30 to 450 °C at a heating rate of 10 °C min−1.Anti-microbial activity
The following microorganisms were used: Micrococcus luteus NCTC 7743, Staphylococcus aureus NCTC 4163, Staphylococcus epidermidis ATCC 49134, Enterococcus faecium ATCC 49474, Moraxella catarrhalis ATCC 25238, Escherichia coli ATCC 25922, Serratia marcescens ATCC 8100, Proteus vulgaris NCTC 4635, Pseudomonas aeruginosa NCTC 6749, Bacillus subtilis ATCC 6633, Candida albicans ATCC 10231, and Rhodothorula rubra (Demml 1889, Lodder 1934). These are the pathogenic microorganisms (bacteria and fungi) used by us for several years. They allow us to effectively evaluate the microbial activity of the synthesized ILs.Anti-microbial activity was determined by the tube dilution method. Bacteria strains were cultured on a Müller–Hinton broth for 24 h and fungi on Sabouraud agar for 48 h. A suspension of the microorganisms at a concentration of 106 cfu mL−1 was prepared from each culture. This suspension was used to inoculate the broth medium, which was then added at ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to each dilution of tested sample prepared in the broth medium. Growth of the microorganisms (or lack thereof) was determined visually after incubation for 24 h at 37 °C (bacteria) or 48 h at 29 °C (fungi).
:1 to each dilution of tested sample prepared in the broth medium. Growth of the microorganisms (or lack thereof) was determined visually after incubation for 24 h at 37 °C (bacteria) or 48 h at 29 °C (fungi).
The lowest concentration at which there was no visible growth (turbidity) was taken as the MIC (minimal inhibitory concentration). Then, an aliquot taken from each tube in a sample loop was cultured in an agar medium with inactivates (0.3% lecithin, 3% polysorbate 80, and 0.1% L-cysteine) and incubated for 48 h at 37 °C (bacteria) or for 5 d at 29 °C (fungi). The lowest concentration of the studied ILs supporting no colony formation was defined as the MBC (minimal bactericidal concentration) or MFC (minimal fungicidal concentration).
Dissolution of cellulose
A vial containing 1.0 g of the dried IL (or a solution of 1.0 g of IL and DMSO in different proportions) was immersed in an oil bath at a given temperature (105 °C). The instability of the bath temperature was estimated to be ±1 °C. Microcrystalline cellulose was added in mass fractions of 0.005 or 0.01 and stirred. Additional cellulose was added after the solution became optically clear under a microscope (Bresser Biolux LCD Microscope). The process was repeated until the cellulose became saturated. This was judged to be the point where cellulose could not be dissolved further.Concentration of cellulose dissolved in IL was calculated from equation 1,
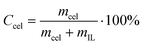 | (1) |
Concentration of cellulose dissolved in IL/DMSO solution was calculated from Formula 2,
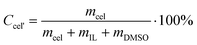 | (2) |
Scanning electron microscopy
Scanning electron microscopy (SEM) observations of cellulose morphology were performed using TESCAN Vega TS 5135 at an acceleration voltage of 1.5–3.0 kV. Carbon films were deposited onto the surface by evaporation.Calculations
The structures of cellobiose dimer were minimized starting with the X-ray structure coordinates obtained from the Cambridge Structural Database (CSD).21 The DFT calculations reported in this study were carried out with Gaussian 09.22 All geometries optimized of “close” and “open” dispositions were computed at the X3LYP level using 6-31+G(d,p) basis set with default loose criteria. To estimate energy changes in the DMSO solution, the default integral equation formalism polarizable continuum model (IEF-PCM)23 with full geometry optimization was employed at the PCM/X3LYP/6-31+G(d,p) level.Results and discussion
Alkyl(cyclohexyl)dimethylammonium bromides were synthesized in the quaternization reaction. The quaternization proceeded via a Menschutkin reaction involving an SN2 mechanism. Quaternary ammonium bromides 1–9, presented in Table 1, were obtained in 24 h at room temperature with very good yield. The purities (surfactant content) of these hydrophilic bromides were determined by a direct two-phase titration technique (EN ISO 2871-2: 2010) and ranged between 98 and 99%.| Salt | R1 | Yield (%) | Mp (°C) | Surfactant content (%) |
|---|---|---|---|---|
| 1 | C2H5 | 95 | 259–260 | – |
| 2 | C4H9 | 95 | 172–174 | – |
| 3 | C6H13 | 94 | 160–162 | – |
| 4 | C8H17 | 96 | 180–182 | 99 |
| 5 | C10H21 | 95 | 163–165 | 98 |
| 6 | C12H25 | 87 | 165–167 | 99 |
| 7 | C14H29 | 84 | 156–157 | 98 |
| 8 | C16H33 | 82 | 129–131 | 98 |
| 9 | C18H37 | 81 | 137–139 | 99 |
Potassium hydroxide was added to alkyl(cyclohexyl)dimethylammonium bromide, dissolved in methanol. KBr was removed by filtering from the reaction mixture. Then, a stoichiometric amount of organic acid was added to the filtrate. The reaction, conducted according to Scheme 1, took place immediately, with high yields. The synthesized formates (Fmt) 10–12, acetates (OAc) 13–18, 2-methoxyacetates (MAc) 19–21, 2-(2-methoxyethoxy)acetates (MEAc) 22–24, 2-[2-(2-methoxyethoxy)ethoxy]acetates (MEEAc) 25–27 and 2-ethoxyacetates (EAc) 28-30 are presented in Table 2. All of the obtained salts were liquids at room temperature, except acetate 13 (solid, mp = 50–51 °C) and five waxes: 10, 22, 28, 29 and 30. The synthesized salts were stable in air as well as in contact with water and popular organic liquids. They were dried in vacuum at 80 °C for 24 h and stored over P4O10. The water content of dried ILs, determined by Karl Fischer measurements, were found to be less than 500 ppm. The obtained ILs were insoluble in hexane and ethyl acetate but were soluble in water, acetone, chloroform, DMSO and low molecular weight alcohols.
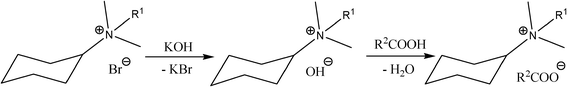 | ||
| Scheme 1 | ||
| IL | R1 | R2 | Abbreviation of anion | Yield (%) | Viscositya (mPa s) | Viscosityb (mPa s) | Density (g mL−1) | Refractive index |
|---|---|---|---|---|---|---|---|---|
| a at 25 °C; b at 100 °C. | ||||||||
| 10 | C4H9 | H | Fmt | 94 | – | 56.3 | – | – |
| 11 | C6H13 | H | Fmt | 95 | 39.9 | 1.5 | 1.03 | 1.486 |
| 12 | C8H17 | H | Fmt | 90 | 168.7 | 2.3 | 1.02 | 1.486 |
| 13 | C2H5 | CH3 | OAc | 98 | – | 0.9 | – | – |
| 14 | C4H9 | CH3 | OAc | 97 | 66.6 | 3.0 | 1.06 | 1.490 |
| 15 | C6H13 | CH3 | OAc | 99 | 100.1 | 1.8 | 1.04 | 1.490 |
| 16 | C8H17 | CH3 | OAc | 98 | 216.7 | 2.5 | 1.02 | 1.490 |
| 17 | C10H21 | CH3 | OAc | 96 | 68.0 | 2.0 | 1.00 | 1.480 |
| 18 | C12H25 | CH3 | OAc | 99 | 27.0 | 5.6 | 0.99 | 1.483 |
| 19 | C4H9 | CH3OCH2 | MAc | 96 | 57.4 | 1.6 | 1.10 | 1.490 |
| 20 | C6H13 | CH3OCH2 | MAc | 97 | 37.8 | 1.7 | 1.07 | 1.491 |
| 21 | C8H17 | CH3OCH2 | MAc | 96 | 56.4 | 1.8 | 1.04 | 1.490 |
| 22 | C4H9 | CH3OCH2CH2OCH2 | MEAc | 97 | – | 0.8 | – | – |
| 23 | C6H13 | CH3OCH2CH2OCH2 | MEAc | 97 | 36.4 | 1.3 | 1.08 | 1.490 |
| 24 | C8H17 | CH3OCH2CH2OCH2 | MEAc | 96 | 41.1 | 1.5 | 1.06 | 1.487 |
| 25 | C4H9 | CH3O(CH2CH2O)2CH2 | MEEAc | 98 | 34.5 | 1.2 | 1.10 | 1.482 |
| 26 | C6H13 | CH3O(CH2CH2O)2CH2 | MEEAc | 98 | 64.7 | 1.7 | 1.09 | 1.481 |
| 27 | C8H17 | CH3O(CH2CH2O)2CH2 | MEEAc | 98 | 142.4 | 2.3 | 1.08 | 1.482 |
| 28 | C4H9 | CH3CH2OCH2 | EAc | 95 | – | 2.7 | – | – |
| 29 | C6H13 | CH3CH2OCH2 | EAc | 90 | – | 2.9 | – | – |
| 30 | C8H17 | CH3CH2OCH2 | EAc | 94 | – | 3.0 | – | – |
Table 2 lists viscosity, density and refractive indices of synthesized ILs. The viscosity of studied ILs at 25 °C varied from 27.0 for 18 to 216.7 mPa s for 16. The values were strongly temperature-dependent. At a temperature of 100 °C the viscosity was low, fitting the range of 0.8 to 5.6 mPa s, except of 10, the viscosity of which amounted to 56.3 mPa s. Densities of the prepared ILs at the temperature of 25 °C ranged between 0.99 and 1.1 g mL−1. For salts with the same anion, their densities decreased in parallel to elongation of the alkyl chain by the quaternary nitrogen atom. On the other hand, elongation of the chain in the anion beginning at Fmt, OAc, MAc, MEAc and ending at MEEAc resulted in an increase in density. Refractive indices measured for all the synthesized ILs ranged within the narrow scope of 1.481 to 1.491.
All of the prepared ILs were characterized by 1H NMR, 13C NMR and elemental analysis. In 1H NMR spectra proton chemical shifts –N+–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–alkyl were observed for the protons located around the quaternary nitrogen atom. The substitution of the bromide anion for the alkylcarboxylate anion caused the chemical shift to decrease by 0.26–0.17 ppm. The 13C NMR spectra of these ILs indicated no significant variation in the carbon signals. Such shifts in proton spectrum between precursor and IL were recorded earlier.24
2–alkyl were observed for the protons located around the quaternary nitrogen atom. The substitution of the bromide anion for the alkylcarboxylate anion caused the chemical shift to decrease by 0.26–0.17 ppm. The 13C NMR spectra of these ILs indicated no significant variation in the carbon signals. Such shifts in proton spectrum between precursor and IL were recorded earlier.24
14N chemical shifts of ammonium cations are sensitive to presence of counterions25 and we decided to apply nitrogen NMR spectroscopy for examination of anion effect and of cellulose presence. 14N NMR spectra recorded for bromides 1–3 showed negligible effect of the alkyl chain or counterion on nitrogen chemical shift, corresponding to similar electron deficiency around nitrogen atoms in the analyzed compounds. The replacement of bromide by acetate anion, as in 3 and 15, causes only 1 ppm shielding. The presence of 17.5% cellulose additive did not change the nitrogen chemical shift of the ammonium cation (−319.74 ppm).
NMR spectra recorded for (cyclohexyl)hexyldimethylammonium acetate (15) at 700 MHz allowed assignment of 1H chemical shifts precisely and signals of cyclohexyl axial and equatorial protons were distinguished. The presence of correlation in the ROESY spectrum between cyclohexyl H-1 and H-2eq protons supports the axial location of the H-1 proton. This suggests the equatorial position of the ammonium nitrogen atom, as already was observed for the crystal cyclohexylammonium salt.26
Recently the effect of cellobiose concentration on the carbon chemical shifts of 1-ethyl-3-methylimidazolium acetate [emim][OAc] in DMSO-d6 was reported.27 Most sensitive to the presence of cellulose were chemical shifts of the C2 carbon atom of the imidazolium cation and both carbon atoms of the acetic anion. Changes of chemical shifts were determined based on the DMSO-d6 signal as an internal signal. Such methodology was questioned by Remsing, as the presence of cellulose can affect the solvent signal position.28 Thus, we decided to analyze chemical shifts of ILs 14 and 15 in 13C spectra recorded in DMSO-d6, additionally using nitromethane-d3 as the external standard. When the solvent was used as an internal standard chemical shifts of all carbon atoms in 14 and 15 were practically unchanged, except the signal of the methyl group in acetic anions in the presence of cellulose (20%). Differences Δδ in the absence and presence of cellulose were smaller than 0.07 ppm and are practically negligible contrary to changes observed by Zhang.27 Chemical shifts of [emim][OAc] anion were shifted about −0.4 ppm (CH3) and +0.9 ppm (COO−) in the presence of 20% cellulose. Analysis of 13C chemical shifts of 14 and 15 related to external standards revealed that chemical shifts of all cation carbon atoms were at the same position (14) or moved 0.30–0.41 ppm (15) (Table 3). For the acetic anion methyl group, the observed cellulose shift was 0.83 ppm. Such an effect can be explained by overall changes of magnetic susceptibility of solution with only small local changes due to IL–cellulose interactions.
| Carbon atom | 14 | 15 | ||||||
|---|---|---|---|---|---|---|---|---|
| 0% | 13% | 0% | 20% | |||||
| δ 0 | Δδa | δ 0 | Δδa | |||||
| CD3NO2 | DMSO-d6 | CD3NO2 | DMSO-d6 | CD3NO2 | DMSO-d6 | CD3NO2 | DMSO-d6 | |
| a Δδ = δ − δ0. | ||||||||
| CH3 | 23.11 | 24.91 | −0.12 | −0.10 | 23.31 | 24.92 | 0.83 | 0.49 |
| COO− | 171.21 | 173.01 | 0.12 | 0.14 | 171.62 | 173.22 | 0.34 | 0.01 |
| CH3–C | 11.86 | 13.66 | −0.02 | 0.00 | 12.36 | 13.97 | 0.35 | 0.01 |
| CH3–N | 45.98 | 47.77 | −0.01 | 0.02 | 46.17 | 47.77 | 0.35 | 0.02 |
| CH | 68.96 | 70.76 | 0.02 | 0.04 | 69.17 | 70.78 | 0.41 | 0.07 |
| (CH2)2′,6′ | 23.68 | 25.48 | −0.03 | −0.01 | 23.90 | 25.51 | 0.31 | −0.03 |
| (CH2)3′,5′ | 23.22 | 25.01 | −0.02 | 0.01 | 23.42 | 25.03 | 0.34 | 0.00 |
| (CH2)4′ | 22.75 | 24.55 | −0.03 | −0.01 | 22.98 | 24.58 | 0.30 | −0.03 |
| CH2-1 | 59.85 | 61.64 | 0.01 | 0.04 | 60.22 | 61.82 | 0.40 | 0.07 |
| CH2-2 | 21.95 | 23.75 | −0.04 | −0.02 | 20.10 | 21.71 | 0.31 | −0.03 |
| CH2-3 | 17.62 | 19.42 | −0.02 | 0.00 | 24.09 | 25.69 | 0.32 | −0.01 |
| CH2-4 | — | — | — | — | 29.25 | 30.85 | 0.31 | −0.02 |
| CH2-5 | — | — | — | — | 20.49 | 22.10 | 0.32 | −0.02 |
The differences were observed in spectra of –N+–C2–alkyl recorded at 700 MHz for bromides 1–3 and acetate 15. Only for ethyl derivative 1, the typical quartet corresponding to coupling with three equivalent protons was observed. For butyl (2) and hexyl (3 and 15) ILs resonances of CH2 protons of attached to nitrogen carbon atoms were recorded as multiplets. The complete analyses of these multiplets was impossible due to relatively poor spectra resolution (half-width of lines about 1 Hz), however, their patterns were recognized as a half spectrum of an AA′XX′ system. The lack of magnetic equivalency of protons suggests slow, in an NMR time scale, rotation around the C1–C2 bond in butyl or hexyl chains.29
For the synthesized salts phase transitions and breakdown temperatures were determined. The results were listed in Table 4. For bromides 2, 3, 5, 6 two melting points (Tm) were observed, one below and the other above 100 °C. The lower temperature was evident in the first cycle, in the second cycle it was reproducible only for bromides 2 and 3. Two melting temperatures pointed to conformation of the cyclohexyl substituent and to an effect of the alkyl group. Only for shorter alkyl groups (from ethyl to octyl) two melting temperatures were noted in the second cycle.
| Salt | Tg (°C)a | Tc (°C)b | Tm (°C)c | Tonset (°C)d |
|---|---|---|---|---|
| a Tg—glass transition temperature.b Tc—crystallization temperature was determined by DSC on heating. c Tm—melting point. d Tonset—decomposition temperature. | ||||
| 1 | – | — | — | 263.0 |
| 2 | – | 151.6 | 63.3/173.1 | 214.1 |
| 3 | – | 83.4/122.5 | 93.8/167.1 | 205.6 |
| 4 | – | 142.1 | 180.6 | 204.0 |
| 5 | – | 137.4 | 62.7/162.8 | 205.9 |
| 6 | – | 136.1 | 70.0/164.5 | 196.9 |
| 10 | −69.70 | −3.70 | – | 183.0 |
| 11 | −63.00 | −17.80 | 22.6 | 177.0 |
| 12 | −56.30 | −0.84 | 56.3 | 182.2 |
| 13 | −80.3 | – | – | 168.0 |
| 14 | −53.22 | 3.74 | 50.37 | 162.2 |
| 15 | −49.47 | – | – | 163.4 |
| 16 | −58.37 | – | – | 160.0 |
| 17 | −71.5 | – | – | 190.8 |
| 18 | −68.8 | – | – | 170.2 |
| 19 | −58.43 | – | – | 178.4 |
| 20 | −57.06 | – | – | 187.0 |
| 21 | −49.11 | – | – | 184.6 |
| 22 | −62.07 | – | – | 172.5 |
| 23 | −60.12 | – | – | 184.7 |
| 24 | −60.77 | – | – | 178.6 |
| 25 | −60.61 | – | – | 174.5 |
| 26 | −65.20 | – | – | 177.5 |
| 27 | −63.80 | – | – | 187.3 |
| 28 | −57.89 | – | – | 172.7 |
| 29 | −57.32 | – | – | 176.4 |
| 30 | −57.97 | – | – | 174.5 |
The results listed in Table 4 demonstrated how strongly the exchange of bromide anion affected the thermal properties of the obtained ILs 10–30. For all ILs the glass transition temperatures are observed in the range from −49 to −80 °C. For bromides the glass transition temperatures were not noticed. A decisive majority of synthesized ILs involved compounds that were liquid between glass transition temperature up to decomposition temperature. In parallel, their decomposition temperature decreased.
Alkyl(cyclohexyl)dimethylammonium bromides 1–9 and acetates 13–18 were tested for anti-microbial activity against rods, cocci and fungi. Values of minimum inhibitory concentration (MIC), minimum bactericidal concentration (MBC) and minimum fungicidal concentration (MFC) were established and are compared in Table 5 for the active salts only (4–9, 16–18) with the corresponding values for the widely applied benzalkonium chloride—BA. The anti-microbial activity manifested by the obtained bromides and acetates depended on the length of their alkyl substituent. The activity was proven for salts with the alkyl substituent containing eight or more carbon atoms. Salts with shorter alkyl substituents proved to be inactive. In cases of the examined salts, the number of carbon atoms in the alkyl group from which the compound began to be active was eight. The most active proved to be (cyclohexyl)hexadecyldimethylammonium bromide (8), which was more effective in its action than benzalkonium chloride, as evident in Fig. 1. In Fig. 1 mean values of MIC for all the studied microbes are presented and, beyond doubt, it showed that exchange of the bromide to acetate anion failed to affect its activity. The observations proved to be consistent with the literature data.30,31
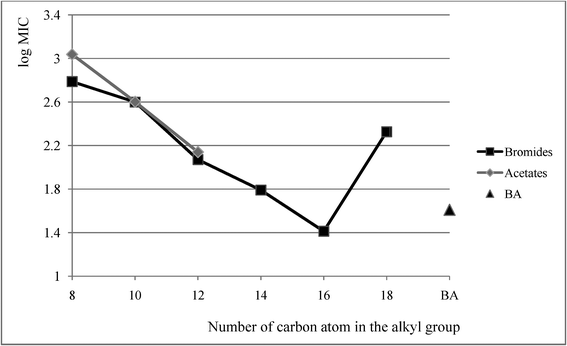 | ||
| Fig. 1 Mean MIC values of bromides 4–9 acetates 16–18 and benzalkonium chloride BA. | ||
| Strain | Salts | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| — | 4 | 5 | 6 | 7 | 8 | 9 | 16 | 17 | 18 | BAb | |
| a In μM. b Benzalkonium chloride (alkyl group represents a mixture ranging from C8H17 to C18H37). | |||||||||||
| Micrococcus luteus | MIC | 6.3 | 46.1 | 10.7 | 2.5 | 2.3 | 2.2 | 26.7 | 5.3 | 2.8 | 1.4 |
| MBC | 194.2 | 178.6 | 10.7 | 2.5 | 4.6 | 2.2 | 1669.5 | 165.1 | 5.6 | 11.4 | |
| Staphylococcus aureus | MIC | 97.1 | 23.0 | 10.7 | 5.0 | 4.6 | 67.5 | 834.8 | 82.6 | 11.2 | 2.9 |
| MBC | 783.2 | 89.3 | 10.7 | 76.9 | 18.5 | 67.5 | 1669.5 | 165.1 | 22.5 | 22.8 | |
| Staphylococcus epidermidis | MIC | 97.1 | 46.1 | 10.7 | 5.0 | 18.5 | 34.8 | 834.8 | 42.6 | 5.6 | 1.4 |
| MBC | 391.6 | 89.3 | 42.6 | 39.7 | 18.5 | 135.0 | 834.8 | 82.6 | 11.2 | 5.7 | |
| Enterococcus faecium | MIC | 783.2 | 89.3 | 10.7 | 5.0 | 4.6 | 34.8 | 1669.5 | 82.6 | 11.2 | 5.7 |
| MBC | 1566.5 | 360.0 | 21.3 | 9.9 | 9.3 | 135.0 | 1669.5 | 665.8 | 22.5 | 22.8 | |
| Moraxella catarhalis | MIC | 97.1 | 46.1 | 10.7 | 5.0 | 9.3 | 17.4 | 1669.5 | 165.1 | 11.2 | 0.6 |
| MBC | 783.2 | 178.6 | 10.7 | 9.9 | 37.1 | 17.4 | 1669.5 | 332.9 | 22.5 | 1.4 | |
| Escherichia coli | MIC | 391.6 | 23.0 | 5.3 | 2.5 | 2.3 | 34.8 | 103.5 | 10.7 | 1.4 | 2.9 |
| MBC | 783.2 | 46.1 | 5.3 | 2.5 | 9.3 | 34.8 | 417.4 | 21.3 | 5.6 | 2.9 | |
| Seratia marcescens | MIC | >1566.5 | >1440.0 | 333.1 | 310.0 | 71.9 | >1088.5 | >1669.5 | 1331.6 | 703.0 | 177.0 |
| MBC | >1566.5 | >1440.0 | 333.1 | 310.0 | 289.8 | >1088.5 | >1669.5 | 1331.6 | 703.0 | 177.0 | |
| Proteus vulgaris | MIC | >1566.5 | >1440.0 | 333.1 | 76.9 | 37.1 | 67.5 | >1669.5 | 1331.6 | 174.4 | 88.5 |
| MBC | >1566.5 | >1440.0 | 333.1 | 153.7 | 71.9 | 135.0 | >1669.5 | 1331.6 | 351.5 | 88.5 | |
| Pseudomonas aeruginosa | MIC | >1566.5 | >1440.0 | 666.2 | 310.0 | 143.7 | >1088.5 | >1669.5 | 1331.6 | 703.0 | 177.0 |
| MBC | >1566.5 | >1440.0 | 666.2 | 310.0 | 289.8 | >1088.5 | >1669.5 | 1331.6 | 703.0 | 177.0 | |
| Bacillus subtilis | MIC | 783.2 | 89.3 | 10.7 | 5.0 | 9.3 | 67.5 | >1669.5 | 165.1 | 11.2 | 2.9 |
| MBC | 783.2 | 178.6 | 10.7 | 9.9 | 9.3 | 67.5 | >1669.5 | 332.9 | 11.2 | 2.9 | |
| Candida albicans | MIC | 194.2 | 46.1 | 5.3 | 9.9 | 4.6 | 34.8 | 417.4 | 165.1 | 11.2 | 11.4 |
| MFC | 391.6 | 89.3 | 10.7 | 19.8 | 37.1 | 67.5 | 834.8 | 165.1 | 45.0 | 88.5 | |
| Rhodotorula rubra | MIC | 194.2 | 46.1 | 5.3 | 5.0 | 2.3 | 8.7 | 834.8 | 82.6 | 11.2 | 22.8 |
| MFC | 194.2 | 89.3 | 10.7 | 9.9 | 2.3 | 8.7 | 1669.5 | 165.1 | 45.0 | 88.5 | |
Except for two acetates, 17 and 18, the synthesized ILs showed the ability to dissolve cellulose. The studies on cellulose solubility in ILs were conducted at a temperature of 100 °C. Solubility of microcrystalline cellulose (MCC) in the studied ILs reflected mainly the size of the cation but the size of the anion was also of significance. As shown in Table 6, MCC solubility was highest in acetates 14–16, followed by solubility in 2-ethoxyacetate 28 and, in turn, 2-[2-(2-methoxyethoxy)ethoxy]acetate 25, 2-(2-methoxyethoxy)acetates 22, 23 and formate 11. These were the salts containing in their cation butyl and hexyl and in the case of salt 16, octyl groups attached to quaternary nitrogen. The estimated biological activity demonstrated that cellulose was dissolved in salts manifesting no anti-bacterial or anti-fungal activity.
| IL | Maximum concentration of cellulose (%)a | Maximum concentration of cellulose (g mol−1(IL)) |
|---|---|---|
| a Calculated from equation 1. | ||
| 10 | 3.5 | 8.03 |
| 11 | 4.5 | 11.58 |
| 12 | 1.0 | 2.85 |
| 13 | 1.0 | 2.15 |
| 14 | 7.5 | 18.25 |
| 15 | 9.0 | 24.43 |
| 16 | 6.0 | 17.97 |
| 19 | 1.0 | 2.73 |
| 20 | 1.5 | 4.52 |
| 21 | 1.0 | 3.30 |
| 22 | 4.5 | 14.29 |
| 23 | 4.5 | 15.55 |
| 24 | 1.5 | 5.63 |
| 25 | 5.5 | 19.88 |
| 26 | 4.0 | 15.58 |
| 27 | 1.5 | 6.26 |
| 28 | 7.0 | 20.12 |
| 29 | 4.0 | 12.62 |
| 30 | 1.0 | 3.44 |
At the end point, the mixture was mainly too viscous to proceed further. This is why we decided to use DMSO to lower the viscosity. For (cyclohexyl)hexyldimethylammonium acetate 15, which evolved to be the best cellulose solvent, studies on the dissolution of cellulose with the addition of DMSO were conducted. Various proportions of DMSO and IL were examined, to determine the most effective one. The maximum cellulose concentrations in the synthesized IL are listed in Table 7. The cellulose concentration in the IL was found to vary depending on the amount of DMSO added, but the experiments showed that it was possible to obtain a high level of dissolved cellulose—22.5%, in certain proportions of IL and DMSO. This level was obtained after addition of 1.75 ÷ 4.00 g DMSO to 1.00 g of IL. The contents of individual liquid components, dependent on the DMSO content in the solution, are presented in Fig. 2.
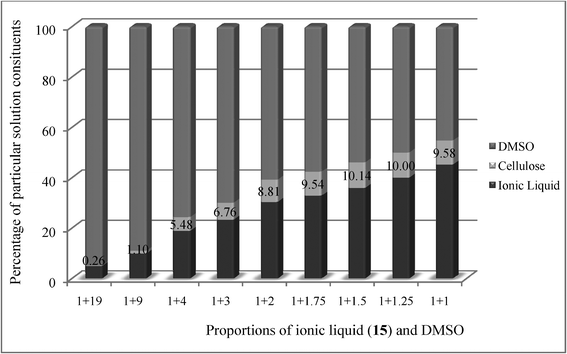 | ||
| Fig. 2 Contents of individual components. The percentage values of cellulose content were calculated using equation 2. | ||
| IL (g) | Cellulose (g) | DMSO (g) | Concentration of cellulosea (%) |
|---|---|---|---|
| a Calculated from equation 2. | |||
| 1.00 | 0.053 | 19.00 | 5.0 |
| 1.00 | 0.111 | 9.00 | 10.0 |
| 1.00 | 0.290 | 4.00 | 22.5 |
| 1.00 | 0.290 | 3.00 | 22.5 |
| 1.00 | 0.290 | 2.00 | 22.5 |
| 1.00 | 0.290 | 1.75 | 22.5 |
| 1.00 | 0.282 | 1.50 | 22.0 |
| 1.00 | 0.250 | 1.25 | 20.0 |
| 1.00 | 0.212 | 1.00 | 17.5 |
Chemical shifts recorded for cellulose are consistent with the data published earlier.32 The concentration was calculated based on a quantitative analysis of two signals at 4.36 (broad singlet of H-1, cellulose) and 0.88 (triplet corresponding to methyl group of the alkyl group). For the solution of acetate 15 and DMSO, in weight ratio 1:1, the integrations were found to originate from deconvolution of signals and the content of cellulose equal to 15.8% was calculated (starting values of 17.5%—Table 7).
MCC solubility in acetate 15 was found to take place at elevated temperatures, beginning at 70 °C. At this temperature, 1% addition of MCC to 15 dissolved fully in 30 min while at temperatures of 80, 90 and 100 °C in, respectively, 25, 15 and 5 min.
A representative morphology of microcrystalline cellulose and cellulose precipitated from a cellulose–acetate 15 solution is shown in Fig. 3. Fig. 3a and 3c present microcrystalline cellulose, Fig. 3b and 3d represent cellulose precipitated by addition of methanol and dried. Fig. 3a and 3b (magnification 500×) demonstrate that cellulose aggregates were larger in the case of precipitated cellulose. The same is shown in Fig. 3c and 3d at a magnification of 3000×. Regeneration changed the morphology of the material: the cellulose became more amorphous than the microcrystalline cellulose. The regenerated cellulose after IL pretreatment showed reduced crystallinity and it can provide better access for cellulolytic enzymes.33 Morphological changes were tracked on SEM photographs.
 | ||
| Fig. 3 SEM images of cellulose (3a, 3c) and cellulose after dissolution in 15 and regeneration with methanol (3b, 3d). | ||
TG curve of microcrystalline cellulose, regenerated cellulose, IL 15 and IL 15 with 8% cellulose are shown in Fig. 4. Cellulose after dissolution in 15 and regeneration with methanol manifested higher thermal stability (curve d) than the original cellulose (curve c). Its breakdown began at 218 °C and manifested a two-stage course.
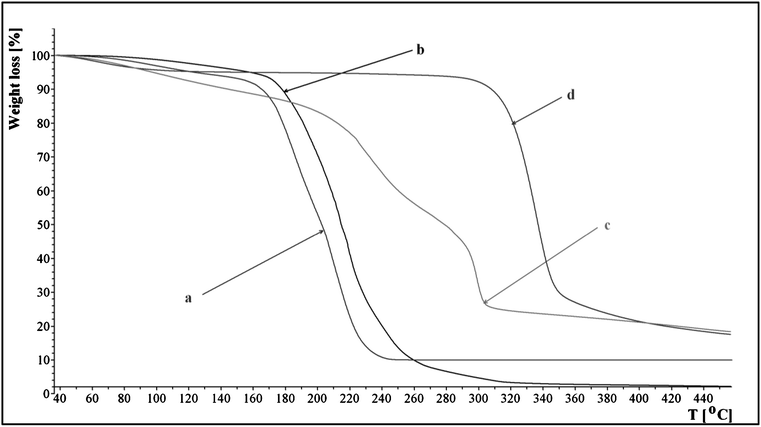 | ||
| Fig. 4 Thermal decomposition profiles of 15, original cellulose and regenerated cellulose: (a) 15, (b) 15 with 8% of microcrystalline cellulose, (c) original cellulose and (d) regenerated cellulose. | ||
In order to study the dissolution process of cellulose we developed a computational model for IL and a cellobiose dimer, which mimics structural relevant, attractive interactions between adjacent polymer chains of MCC fibrils. The crystallographic structure of a cellobiose unit was used as the initial assumption of the dimer formation.21 Two disaccharide units were linked by two O–H⋯O–H bonds, reflecting structural properties of extended zig-zag hydrogen bond pattern of cellulose. Fig. 5 shows association of cellobiose units with (cyclohexyl)hexyldimethylammonium acetate in “open” (5a) and “closed” (5b) disposition. Hydrogen bonds were shown as broken lines, but some C–H hydrogen bonds of ammonium cations and cellobiose were omitted for clarity.
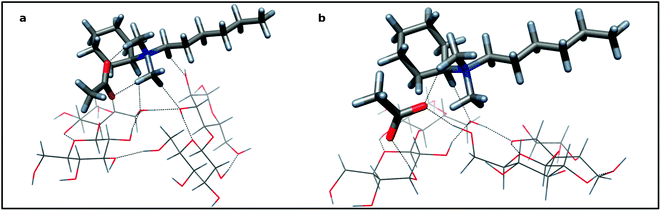 | ||
| Fig. 5 The associate of two cellobiose units and (cyclohexyl)hexyldimethylammonium acetate: 5a—the “closed” disposition of aligned cellobiose dimer (sticks) and (cyclohexyl)hexyldimethylammonium acetate (tubes) as an ionic pair connected with hydrogen bonds, 5b—the “open” disposition with acetate inserted into cellobiose dimer with two sugar residues—acetate hydrogen bonds. Colors of atoms: dark gray—carbon; red—oxygen; blue—nitrogen; light gray—hydrogen. | ||
Structurally, the association of IL molecule to the carbohydrate dimer is symmetrical: the cation prefers the central position (Fig. 5). Presumably, the driving force of the association is the proton donation of the activated C–H bonds of the alkyl groups adjacent to electron with drawing ammonium nitrogen atom (vide supra NMR data). Four hydrogen bonds (ca 2.5–3.1 Å) are directed to the oxygen atoms of both glucose residues of the cellobiose moiety (Fig. 5a). The electrostatic effect and the three other C–H proton donors hold together the cation and anion. Hydrogen bonds to the carboxylate oxygen atoms (ca 2.0 Å) provide considerable stabilization of this ionic pair. The complex formation makes only little strengthening of hydrogen bonds of a cellulose zipper motif: a shortening and improving linearity were observed. Acetate is not in close proximity to the surface of cellobiose dimer and is unable to affect its hydrogen bonds appreciably. However, if we accept the micro-Brownian motions of a segment of cellulose chain and/or some spatial variation of associate components, then the possible short contact of acetate and interchain hydrogen bond is expected and its insertion should be really predicted. Now, the proton donor ability of two hydroxyl groups of the broken bridge may fully exploit proton acceptability of carboxylate. Indeed, two, short, near linear hydrogen bonds are formed (Fig. 5b). The distance between oxygen atoms of participating hydroxyls increases from 2.77 to 4.03 Å, making the rebuilding process of the disrupted linkage almost impossible. The ammonium cation, still linked to its counterion, conserves its position at the center of the cellobiose dimer. At present, the undisturbed, hydrogen bond between two cellobioses weakens, although the ammonium cation is linked with them. Importantly, the whole “open” structure formed is ca 4.2 kcal mol−1 more stable than that preceding “closed” one suggesting that the dissolution process is thermodynamically feasible. The polarizable continuum model (PCM) framework, which allows a more realistic prediction of the DMSO solvation clearly support this conclusion estimating the stabilization energy to be 6.0 kcal mol−1. It is worth noting that sodium acetate (as a carboxylate) is well tailored to effective hydrogen bonding with the vicinal diols of the cellulose unit. We calculate that for β–methyl 4-O-methylglucoside the interaction energy with 2,3-diol is of −36.9 kcal mol−1 in vacuum (but with PCM, still −15.1 kcal mol−1). For comparison, two DMSO molecules interact with this methylated glucoside with energy −21.9 kcal mol−1 (PCM gives −12.3 kcal mol−1). Therefore, DMSO as a solvent, seems to be weaker, but still an acetate competitor.
Generally, it seems that the process of cellulose solvation relay on the hydrophobization of its hydroxyl rich polymer first by acetate and DMSO polar molecules and second by tetraalkylammonium ion. Note that, because of considerable sodium acetate concentration, bulk DMSO molecules, at least in part, form aggregates34,35 acting as O-terminal ligands with methyl groups pointing outward from solvated sodium and thus are hydrophobic for the solute. If so, then it appears that the tetraalkylammonium ionic liquid with DMSO as co-solvent and sodium acetate as an active agent constitute the cellulose solvent triade.
Conclusions
We synthesized alkyl(cyclohexyl)dimethylammonium bromides, formates, and acetates and assessed their ability to dissolve cellulose. Cellulose solubility was found to be determined by the type of cation and the type of the anion. Bromides were unable to dissolve cellulose while formates, acetates, 2-methoxyacetates, 2-(2-methoxyethoxy)acetates, 2-[2-(2-methoxyethoxy)ethoxy]acetates, and 2-ethoxyacetates dissolved cellulose. For alkyl(cyclohexyl)dimethylammonium, cation dimensions of alkyl groups were essential for cellulose solubility and were generally limited to butyl, hexyl and octyl as effective substituents. In parallel, the alkyl group was decisive for anti-microbial activity of the entire molecule. The biological anti-microbial activity of a molecule began when length of the alkyl substituent began to block solubility of cellulose.It appears that the high performance of (cyclohexyl)hexyldimethylammonium acetate was an effect of the fine-tunning of all alkyl groups. According to the presented mechanism, two categories of activity can be clearly distinguished: two methyl groups, with low steric hindrance, may use its six activated C–H bonds to link with both the acetate and cellulose surface. In the second category hexyl and especially cyclohexyl (with its low barrier chair-boat interconversion) are symmetry breaking substituents.36 On the one hand, both isomeric hexyl groups are good enough to warrant the solubility of quaternary cations— cellulose hydrophobized chain aggregates in the bulk ionic liquid—sodium organized DMSO phase. On the other hand, hexyls do not show an excessive size to increase viscosity or to seriously hamper the direct interaction of ammonium and cellulose partners. Finally, we show that the role of the ammonium cation is to transport acetate anion from solution to the cellulose surface environment and, thus, to affect hydrogen bond bridges, which make regular linkages of cellulose chains. Our tentative model shows that their disruption and the insertion of the acetate, leads to further, local reorientation of cellobiose residue. This twist increases disorder of the polymer chains, imparting their solubility. It appears that both acetate and ubiquitous, polar DMSO molecules effectively solvate and, thus, isolate frontier hydroxyl groups and prevent the reproduction of former cellulose polymer. However, it should be emphasised that the presented gas-phase model is only tentative because it suffers from the lack of both: bulk solvation interactions and a low degree of polymerization of cellulose. For the validation of the model this should be considered and further refinement is needed.
NMR experiments provided evidence that the ammonium cation occupies the equatorial position at a cyclohexyl ring and its positive charge is not sensitive to length of alkyl chain or counterion type. The flexibility of alkyl chains decreased for butyl or longer substituents.
In solutions its 1H and 13C NMR spectra were identical with those documented by literature data. Cellulose, after dissolution in (cyclohexyl)hexyldimethylammonium acetate and regeneration with methanol, has reduced crystallinity and manifests lower thermal stability.
Acknowledgements
This work was supported by project POIG.01.03.01-30-074/08 and by grant No. 7548/B/H03/2011/40 (National Science Centre, Poland)References
- A. Stark and K. R. Seddon, Kirk-Othmer Encyclopaedia of Chemical Technology, ed. E-mail: A. Seidel, John Wiley & Sons, Inc., New Jersey, 2007, 26, 836-920 Search PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef CAS.
- M. Petkovic and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 3508–3576 Search PubMed.
- W. L. Hough, M. Smiglak, H. Rodriguez, R. P. Swatloski, S. K. Spear, D. T. Daly, J. Pernak, J. E. Grisel, R. D. Carliss, D. M. Soutullo, J. H. Davis and R. D. Rogers, New J. Chem., 2007, 31, 1429–1436 RSC.
- W. L. Hough and R. D. Rogers, Bull. Chem. Soc. Jpn., 2007, 80, 2262–2269 CrossRef CAS.
- J. Pernak, A. Syguda, D. Janiszewska, K. Materna and T. Praczyk, Tetrahedron, 2011, 67, 4838–4844 CrossRef CAS.
- T. Praczyk, P. Kardasz, E. Jakubiak, A. Syguda, K. Materna and J. Pernak, Weed Sci., 2012, 60, 189–192 CrossRef CAS.
- J. Pernak, A. Syguda, K. Materna, E. Janus, P. Kardasz and T. Praczyk, Tetrahedron, 2012, 68, 4267–4273 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D.Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J. P. Mikkoa, Ind. Crops Prod., 2010, 32, 175–201 CrossRef.
- N. Sun, H. Rodriguez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47, 1405–1421 RSC.
- A. D. Fort, R. P. Swatloski, P. Moyna and G. Moyna, Chem. Commun., 2006, 714–716 Search PubMed.
- H. Zhao, C. L. Jones, G. A. Baker, S. Xia, O. Olubajo and V. N. Person, J. Biotechnol., 2009, 139, 47–54 CrossRef CAS.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS.
- O. A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn and T. Heinze, Biomacromolecules, 2007, 8, 2629–2647 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- E. Kalenius, T. Kekalainen, R. Neitola, K. Beyeh, K. Rissanen and P. Vainiotalo, Chem.–Eur. J., 2008, 14, 5220 CrossRef CAS . Cambridge Structural Database (CSD), entry code: CELLOB 04.
- Gaussian 09, Revision A.02 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef.
- J. Pernak, A. Syguda, I. Mirska, A. Pernak, J. Nawrot, A. Prądzyńska, S. T. Griffin and R. D. Rogers, Chem.–Eur. J., 2007, 13, 6817–6827 CrossRef CAS.
- M. Witanowski, L. Stefaniak and G. A. Webb, Ann. R. NMR S., 1986, 18, 90–94 Search PubMed.
- T. Sakano, M. Horie, K. Osakade and H. Nokao, Bull. Chem. Soc. Jpn., 2001, 74, 2059–2065 CrossRef CAS.
- J. Zhang, H. Zhang, J. Wu, J. Zhang, J. He and J. Xiang, Phys. Chem. Chem. Phys., 2010, 12, 1941–1947 RSC.
- R. C. Remsing, I. D. Petrik, Z. Liu and G. Moyna, Phys. Chem. Chem. Phys., 2010, 12, 14827–1428 RSC.
- E. W. Garbisch Jr, J. Chem. Educ., 1968, 45, 480–493 CrossRef.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- J. Pernak, K. Sobaszkiewicz and I. Mirska, Green Chem., 2003, 5, 52–56 RSC.
- S. Hesse-Ertelt, T. Heinze, B. Kosan, K. Schwikal and F. Meister, Macromol. Symp., 2010, 294(2), 75–89 CrossRef CAS.
- A. P. Dadi, S. Varanasi and C. A. Schall, Biotechnol. Bioeng., 2006, 95, 904–910 CrossRef CAS.
- M. Calligaris, Coord. Chem. Rev., 2004, 248, 351–375 CrossRef CAS.
- M. Calligaris and O. Carugo, Coord. Chem. Rev., 1996, 153, 83–154 CrossRef CAS.
- I. Lopez-Martin, E. Burello, P. N. Davey, K. R. Seddon and G. Rothenberg, ChemPhysChem, 2007, 8, 690–695 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21502k/ |
| This journal is © The Royal Society of Chemistry 2012 |