Palladium-catalyzed cyanide metathesis: utilization of benzyl cyanide as an operator-benign reagent for aryl halide cyanations†
Qiaodong
Wen
,
Jisong
Jin
,
Binbin
Hu
,
Ping
Lu
* and
Yanguang
Wang
*
Department of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: pinglu@zju.edu.cn; orgwyg@zju.edu.cn; Fax: +86 571-87952543; Tel: +86 571-87952543
First published on 25th June 2012
Abstract
Benzyl cyanide was disclosed to be a safe and effective cyanation reagent for palladium-catalyzed cyanation of haloarenes, which afforded aryl nitriles in moderate to good yields.
Aryl nitriles are important scaffolds in natural and synthetic organic compounds, such as pharmaceuticals, herbicides, agrochemicals, and optoelctronic materials.1 Moreover, transformations of the cyano group into other functionalities have been widely utilized to construct organic molecules of diversity and complexity. Conventional preparative methods toward aryl nitriles are the diazotization of anilines followed by the subsequent Sandmeyer reaction2 and the Rosenmund–von Braun reaction.3 Since then, a series of cyanation reactions have been discovered by using various cyano sources (Scheme 1), including electrophilic cyano (CN+)4 and nucleophilic cyanide (CN−). Apart from these commonly used cyanating reagents, several systems have also been developed by integrating individual C and N sources into a cyano group.5 Among these cyanation methods, Pd-,6 Cu-,7 and Ni-catalyzed8 aromatic cyanations using cyanides are the most commonly used protocols in modern organic chemistry. Such protocols disclose various cyanide sources, including KCN,9 NaCN,10 Zn(CN)2,11 TMSCN,12n-Bu3SnCN,13 CH3CN,14 and (CH3)2CCN(OH).15 Although widely employed, the protocol still suffers from significant drawbacks: (a) the high cyanide-metal affinity which can poison the transition metals and (b) the severe cyanide hazard and the related tedious protection. To overcome the cyanide hazard, Beller and co-workers disclosed nontoxic K4[Fe(CN)6] as a robust CN− source in Pd-catalyzed aryl cyanations.16 This reagent was further extended to many cyanating systems and showed satisfactory functional group compatibility.17 However, notwithstanding its lower toxicity compared with other cyanides, the low solubility of K4[Fe(CN)6] in organic solvents significantly diminishes its applicability as a viable cyanating reagent.17f Herein, we would like to disclose benzyl cyanide as a versatile and operator-benign cyanating reagent in the Pd-catalyzed aryl cyanation reaction.
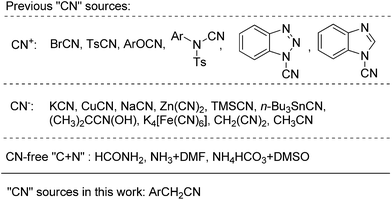 | ||
| Scheme 1 Various types of “CN” sources in preparation of aryl cyanides. | ||
Our initial trial was conducted between 2-iodoaniline and benzyl cyanide with n-Bu4NBr, K2CO3, and a catalytic amount of Pd(OAc)2 in 5 mL DMF. The reaction afforded 2-cyanoaniline in 76% isolated yield. Enlightened by this result, we further optimized the reaction conditions using 9-bromoanthracene as a substrate and benzyl cyanide as the cyanide source. The results are summarized in Table S1 (ESI†).
Firstly, palladium sources were screened. It was shown that the cyanation could be achieved by either PdCl2 or Pd(OAc)2. Using triphenylphosphine-ligated catalyst PdCl2(PPh3)2 or a combination of Pd(OAc)2 and PPh3, 9-bromoanthracene was mostly recovered. Similarly, 9-bromoanthracene was also recovered in 91% when using Pd(PPh3)4 as a catalyst. Gradually varying the amount of Pd(OAc)2 from 2.5 mol% to 10 mol%, we found that the optimal result was obtained with suitable 5 mol% of Pd(OAc)2. Both K2CO3 and n-Bu4NBr were essential. By decreasing the amount of K2CO3 from 5 to 1 equivalents to aryl bromide, a gradually decreased yield was observed. In the absence of K2CO3, the yield decreased from 88% to 40%. When NaHCO3 was used instead of K2CO3, the yield also decreased. Addition of NaOAc resulted in the quantitative recovery of 9-bromoanthracene. When Et3N was used, 9-bromoanthracene was recovered in 77% yield along with 5% of anthracene. Altering the amount of n-Bu4NBr from 0.5 equivalents to 2, the optimal yield was obtained with 1 equivalent of n-Bu4NBr. n-Bu4NCl or n-Bu4NI could also promote the reaction, but with slightly lower yields. In the case where n-Bu4NCl was used, anthracene was isolated in 5% yield except for the formation of the 9-cyanoanthracene. In the presence of n-Bu4NF·3H2O, 79% of 9-bromoanthracene was recovered. Without using K2CO3 and n-Bu4NBr, only a trace amount of 9-cyanoanthracene was detected by TLC, while the 9-bromoanthracene was recovered in 59% yield. By altering the solvent from DMF to dimethylacetamide (DMAc), and dioxane, 9-cyanoanthracene was isolated in 34% and 54% yields, while 46% and 30% of aryl bromide was recovered, respectively. When DMSO was used, the cyanation product was not detected, and 93% of 9-bromoanthracene was recovered. A similar situation was observed in THF. Finally, a suitable reaction temperature and reaction time were found to be 90 °C and 8 h, respectively. Thus, the optimal reaction conditions were established. When we changed benzyl cyanide into 4-fluorobenzyl cyanide, the yield of 9-cyanoanthracene was significantly improved to 94%. 4-Methoxybenzyl cyanide afforded the desired product in 51% yield (Scheme 2).
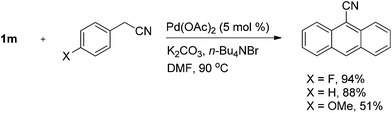 | ||
| Scheme 2 Effect of substituents of benzyl cyanides on the Pd-catalyzed cyanation of 9-bromoanthracene. | ||
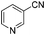
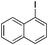
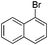
With the optimized reaction conditions in hand, we tested the substrate diversity. With the electron donating groups (NH2, CH3O, CH3) at the para position of the iodobenzene, the corresponding aryl cyanides were obtained in 66–80% yields (Table 1, entries 2–4). When 4-iodotoluene was subjected to the reaction, 4,4′-dimethyl-1,1′-biphenyl was isolated in 20% yield along with 4-cyanotoluene in 66% yield. 4-Iodophenol can also participate in the reaction; however, the desired cyanide was afforded only in 38% yield (Table 1, entry 5). 4-Bromoanisole gave the product in moderate yield (53%) (Table 1, entry 6). Compared with 4-iodoanisole, the lower yield is possibly due to the higher C–Br bond energy than the C–I bond energy. This can be in part confirmed by the fact that 2-chloroaniline and 4-fluoroaniline did not undergo the reaction. When we changed the para-substituent from electron donating groups to electron withdrawing groups, 4-bromoacetophenone gave 4-cyanoacetophenone in 41% yield (Table 1, entry 8). 3-Bromopyridine could also be converted into 3-cyanopyridine, which might be used as the precursor of bioactive compounds (Table 1, entry 10). In contrast, the reaction proceeded well for the fused aryl halides (Table 1, entries 11–15). 1-Cyanonaphthalene could be prepared either from 1-iodonaphthalene or from 1-bromonaphthalene in 75% and 68% yields, respectively. 9,9-Dibutyl-2-iodo-9H-fluorene furnished the corresponding cyanide in 75% yield, while 9-heptyl-3,6-diiodo-9H-carbazole gave dicyanated and monocyanated products in 51% and 12% yields, respectively.
|
|
||||
|---|---|---|---|---|
| Entry | 1 | ArX (1) | ArCN (2) | Yield(%)b |
| a Reaction conditions: substrate (1 mmol), benzyl cyanide (1.5 mmol), K2CO3 (5 mmol), n-Bu4NBr (1 mmol), Pd(OAc)2 (5 mol%), DMF (5 mL), 90 °C (oil bath), 8 h. b Isolated yield. c 3 mmol of benzyl cyanide was used, monocyanated product 2m was isolated in 12% yield. | ||||
| 1 | 1a | 2-NH2C6H4I | 2-NH2C6H4CN | 2a (76) |
| 2 | 1b | 4-NH2C6H4I | 4-NH2C6H4CN | 2b (70) |
| 3 | 1c | 4-CH3OC6H4I | 4-CH3OC6H4CN | 2c (80) |
| 4 | 1d | 4-CH3C6H4I | 4-CH3C6H4CN | 2d (66) |
| 5 | 1e | 4-HOC6H4I | 4-HOC6H4CN | 2e (38) |
| 6 | 1f | 4-CH3OC6H4Br | 4-CH3OC6H4CN | 2c (53) |
| 7 | 1g | 4-PhCOC6H4I | 4-PhCOC6H4CN | 2f (68) |
| 8 | 1h | 4-CH3COC6H4Br | 4-CH3COC6H4CN | 2g (41) |
| 9 | 1i | 4-PhCOC6H4Br | 4-PhCOC6H4CN | 2f (48) |
| 10 | 1j |
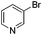
|
|
2h (57) |
| 11 | 1k |
|
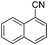
|
2i (75) |
| 12 | 1l |
|
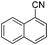
|
2i (68) |
| 13 | 1m |
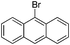
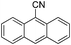
|
|
2j (88) |
| 14 | 1n |
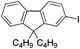
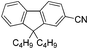
|
|
2k (75) |
| 15 | 1o |
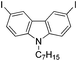
|
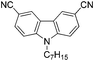
|
2l (51)c |
This protocol could be carried in gram scale. When 1.17 g 1c (5 mmol) was used under the same reaction condition, 2c was obtained in 52% yield although the homocoupling product (4,4′-dimethoxy-1,1′-biphenyl) was formed and isolated in 42% yield. As the importance of 2na in the manufacture of Sartan's medicine, 2-bromo-4′-methyl-1,1′-biphenyl (1p) (5 mmol) was tested as the substrate. A mixture of 2na and 2nb was obtained in 46% yield. The ratio of 2na and 2nb was determined to be about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by GC-MS and further confirmed by 1H NMR. 2na was formed by direct palladium catalyzed cyanation of Ar–X, while 2nb was prepared through 1,4-palladium migration18 between A and Bvia C–H activation (Scheme 3).
:1 by GC-MS and further confirmed by 1H NMR. 2na was formed by direct palladium catalyzed cyanation of Ar–X, while 2nb was prepared through 1,4-palladium migration18 between A and Bvia C–H activation (Scheme 3).
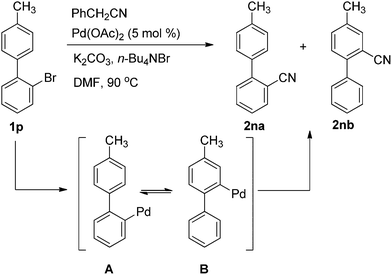 | ||
| Scheme 3 Reaction of 1p with benzyl cyanide. | ||
Based on the screening of reaction conditions and the survey of the substrate diversity, we postulated a possible mechanism for this transformation (Scheme 4). The typical palladium catalyzed cyanation reaction includes three steps, the oxidative addition (step 1), the halide and cyanide exchange, and the reductive elimination (step 4). In the present reaction, the catalytic cycle was expanded. After the usual oxidative addition (step 1), ligand exchange occurred between benzyl cyanide and the halide due to lone pair electrons on the cyanide nitrogen (step 2).19 In this step, a key intermediate C was obtained. Assisted by the nucleophilic attack on the benzylic carbon with the halide, the benzyl halide could be released from the complex. Meanwhile, the palladium migrated from nitrogen to the adjacent carbon atom leading to the formation of ArPdCN (step 3). Finally, the aryl cyanide was obtained via the reductive elimination (step 4). Only a trace amount of cyanide anion was detected20 which was inevitably released from the ArPdCN. Therefore, the poisoning species,21 such as [Pd(CN)4]2−, [ArPd(CN)3]2−, [LPd(CN)3]2−, and [HPd(CN)3]2− were effectively limited. Moreover, because of the coordination of the benzyl cyanide and the palladium, the C–CN bond in PhCH2–CN was activated, which made the nucleophilic substitution on the benzylic carbon possible. The GC-MS analysis of the reaction mixture revealed the existence of benzyl alcohol, which indicated the possible formation of benzyl halides in the catalytic cycle.
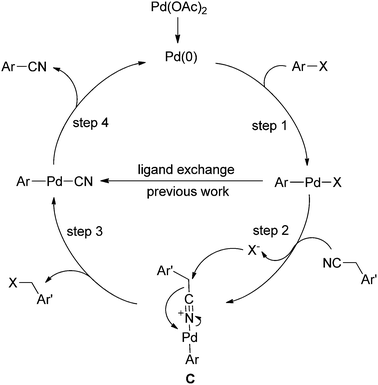 | ||
| Scheme 4 Proposed mechanism for Pd-catalyzed cyanation using benzyl cyanide. | ||
In conclusion, we have developed a practical palladium-catalyzed cyanation of aryl halides using benzyl cyanide as cyanation reagent. The present method has several advantages over the conventional protocols. First, commercially available benzyl cyanide is a cyanide anion-free reagent, which is essentially hazardless and organic solvent soluble. Second, the poison of the palladium catalysts can be effectively diminished due to the avoidance of the inhibited [Pd(CN)4]2−, [ArPd(CN)3]2−, and [LPd(CN)3]2−. Third, the reaction can be conducted under open-air conditions. Fourth, this is a ligand-free transition metal-catalyzed reaction. Fifth, the protocol can be carried out on a gram scale. Further research on cyanations using benzyl cyanide is currently underway.
Acknowledgements
Ping Lu and Yanguang Wang thank the national nature science foundation of China (20972137, 21032005).References
- M. H. Gebs and R. H. Abeles, J. Med. Chem., 1986, 29, 585 CrossRef
.
- T. Sandmeyer, Eur. J. Inorg. Chem., 1884, 17, 2650 Search PubMed
-
(a) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 52, 1749 Search PubMed
-
(a) K. Buttke and H. J. Niclas, J. Prakt. Chem., 1996, 338, 681 CrossRef CAS
-
(a) J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS
-
(a) X. Jia, D. Yang, W. Wang, F. Luo and J. Cheng, J. Org. Chem., 2009, 74, 9470 CrossRef CAS
-
(a) H. J. Cristau, A. Ouali, J. F. Spindler and M. Taillefer, Chem.–Eur. J., 2005, 11, 2483 CrossRef CAS
- T. Arai, Y. Suemitsu and Y. Ikematsu, Org. Lett., 2009, 11, 333 CrossRef CAS
-
(a) M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem.–Eur. J., 2003, 9, 1828 CrossRef CAS
-
(a) J. R. Dalton and S. L. Regen, J. Org. Chem., 1979, 44, 4443 CrossRef CAS
-
(a) H. Kubota and K. C. Rice, Tetrahedron Lett., 1998, 39, 2907 CrossRef CAS
-
(a) N. Chatani and T. Hanafusa, J. Org. Chem., 1986, 51, 4714 CrossRef CAS
- V. Nair, D. F. Purdy and T. B. Sells, J. Chem. Soc., Chem. Commun., 1989, 878 RSC
- F. Luo, C. Chu and C. Cheng, Organometallics, 1998, 17, 1025 CrossRef CAS
- M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 1661 CrossRef CAS
-
(a) T. Schareina, A. Zapf and M. Beller, Chem. Commun., 2004, 1388 RSC
-
(a) S. A. Weissman, D. Zewge and C. Chen, J. Org. Chem., 2005, 70, 1508 CrossRef CAS
-
(a) M. A. Campo and R. C. Larock, J. Am. Chem. Soc., 2002, 124, 14326 CrossRef CAS
- B. Zhao and X. Lu, Org. Lett., 2006, 8, 5987 CrossRef CAS
-
(a) Detailed results for testing the cyanide was presented in Table S2 (ESI†). J. Kim, J. Choi, K. Shin and S. Chang, J. Am. Chem. Soc., 2012, 134, 2528 CrossRef CAS
- S. Erhardt, V. V. Grushin, A. H. Kilpatrick, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2008, 130, 4828 CrossRef CAS
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental details, optimization of reaction conditions, characterizations, and copies of 1H and 13C NMR spectra for all products. See DOI: 10.1039/c2ra20770b/ |
| This journal is © The Royal Society of Chemistry 2012 |