Partial oxidation of aromatic alcohols via TiO2 photocatalysis: the influence of substituent groups on the activity and selectivity†
Sedat
Yurdakal
*a and
Vincenzo
Augugliaro
*b
aKimya Bölümü, Fen-Edebiyat Fakültesi, Afyon Kocatepe Üniversitesi, Ahmet Necdet Sezer Kampüsü, 03200, Afyonkarahisar, Turkey. E-mail: sedatyurdakal@gmail.com; Fax: +90 272 228 12 35; Tel: +90 272 228 1339/230
b“Schiavello-Grillone” Photocatalysis Group, Dipartimento di Ingegneria Elettrica, Elettronica e delle Telecomunicazioni, di tecnologie Chimiche, Automatica e modelli Matematici (DIEETCAM), University of Palermo, Viale delle Scienze, 90128 Palermo, Italy. E-mail: vincenzo.augugliaro@unipa.it
First published on 16th July 2012
Abstract
Aromatic alcohols with substituent groups in different positions have been partially oxidised to the corresponding aldehydes in a photocatalytic system in order to investigate the influence of the substituents on reactivity and selectivity to aldehyde. To this aim benzyl alcohol, 2-methoxybenzyl alcohol, 3-methoxybenzyl alcohol, 4-methoxybenzyl alcohol, 2,4-dimethoxybenzyl alcohol, 4-hydroxybenzyl alcohol and 4-hydroxy-3-methoxybenzyl alcohol have been photocatalytically oxidised to their corresponding aldehydes in aqueous TiO2 suspensions under near-UV irradiation. Home-made and commercial rutile TiO2 samples were used as photocatalysts. The catalysts were characterized by XRD, BET, SEM, TEM and TGA measurements. For all the used substrates the main oxidation products were the corresponding aldehydes and CO2. The aromatic alcohols showed selectivity values decreasing with the substituent position on the aromatic ring according the following order: para > ortho > meta. In the presence of two substituent groups, the overall oxidation rate increased while the selectivity decreased. The home-made catalyst generally showed selectivity higher but activity lower than those of the commercial one. The results showed that the reaction rate and selectivity were dependent not only on the catalyst properties such as crystallinity and hydrophilicity but also on the kind and position of the substituent groups of the aromatic alcohols.
1 Introduction
For the degradation of pollutants in water and air the ideal photocatalyst should be a stable, inexpensive, non-toxic and, most importantly, highly photocatalytically active material. Several semiconductors cover most of the criteria for an optimal photocatalyst, but TiO2 is the most investigated semiconductor due to its promising photocatalytic activity and stability.1–3 Since the photocatalytic reactions involve hydroxyl radicals, until a few years ago this technology has been considered greatly unselective (especially in water)2,3 and therefore mainly suitable for water and air remediation.4,5Photocatalytic reactions for synthetic purposes are not as common as degradation ones but they have been mainly carried out either in organic solvents or in the gas phase.6–9 However, hydrocarbon oxidation,10 aromatic hydroxylation,11 naphthalene oxygenation,12 heterocyclic functionalisation13 and cyclisation of amino acids14 are some examples of synthetic reactions carried out by photocatalytic systems in water.
One of the main goals of current research on organic syntheses is that of eliminating the environmentally harmful conditions of industrial processes. Among them a very important role is played by the selective oxidation of hydroxyl groups to carbonyl ones. This industrial process is generally performed in organic solvents at high temperature and pressure by using chromate and permanganate ions as stoichiometric oxygen donors; these process conditions are not only expensive in terms of energy and chemicals but also produce dangerous wastes.15,16
Photocatalytic alcohol oxidation has been carried out either in acetonitrile17,18 or in air19 but partial oxidation of aromatic alcohols to the corresponding aldehydes has only been recently carried out in aqueous suspensions of home-prepared anatase,20–22 brookite23,24 and rutile TiO225–27 in laboratory batch reactors and also in continuous reactors.28,29 The partial oxidation of benzyl alcohol, 4-methoxybenzyl alcohol, 4-methylbenzyl alcohol and 4-nitrobenzyl alcohol to the corresponding aldehydes was achieved with both home-prepared (HP) and commercial TiO2 samples, and the influence of the substituent group kind on the reaction rate was studied, this effect being well described by the Hammett's relationship.27 Generally, HP photocatalysts showed selectivity values for aldehyde production far higher than those of commercial TiO2 catalysts.
The textural, bulk and surface properties of HP and commercial catalysts have been investigated21,22 in order to explain their different photocatalytic performances. The textural and intrinsic electronic features of catalysts have been found to be almost the same; on the contrary ATR-FTIR studies indicated that the lower crystallinity and the higher surface hydroxyl group density of HP catalysts with respect to commercial ones may justify the higher selectivity exhibited by HP samples. Both of these properties induce an enhanced hydrophilicity of the TiO2 surface, thus promoting desorption of the photo-produced aldehyde so that its further oxidation is hindered.
On the basis of the above reported results, rutile TiO2 catalyst was home prepared at room temperature in order to obtain a catalyst with a more hydrophilic surface. That catalyst has been used in the present work, which has been devoted to a systematic investigation of the photocatalytic oxidation of benzyl alcohol to the corresponding aldehyde and on the influence of methoxy and hydroxyl groups in ortho, meta and para positions on the performance of the photocatalytic oxidation process. Therefore, benzyl alcohol (BA), 2-methoxybenzyl alcohol (2-MBA), 3-methoxybenzyl alcohol (3-MBA), 4-methoxybenzyl alcohol (4-MBA), 2,4-dimethoxybenzyl alcohol (2,4-DMBA), 4-hydroxybenzyl alcohol (4-HBA) and 4-hydroxy-3-methoxybenzyl alcohol (4-H-3-MBA) have been photocatalytically oxidised to their corresponding aldehydes in water and under near-UV irradiation. For comparison purposes a commercial rutile TiO2 has also been tested in the present research.
2 Experimental
2.1 Catalyst preparation and characterization
The precursor solution was obtained by adding 20 mL of TiCl4 (> 97%, Fluka) to 1000 mL of water contained in a volumetric flask (2 L). At the end of the addition, the resulting solution was stirred for 2 min by a magnetic stirrer and then the flask was sealed and maintained at room temperature (ca. 298 K) for a total aging time of 6 days. After ca. 12 h of aging, the sol became almost transparent and then, after waiting a few (2 or 3) days, the precipitation process started. The solid powder, precipitated at the end of the whole treatment, was separated by centrifugation (20 min at 5000 rpm) and dialysed several times with deionised water until a neutral pH was reached. Then, the sample was again centrifuged and dried at room temperature. The final home-prepared catalyst is hereafter indicated as HPRT. The commercial rutile TiO2 (Sigma Aldrich), indicated as SA, was used as received.X-Ray diffractometry (XRD) patterns of the powders were recorded by a Philips diffractometer (operating at a voltage of 40 kV and a current of 30 mA) using Cu Kα radiation and a 2θ scan rate of 1.28° min−1. The crystallinity of HPRT and SA samples was evaluated following the procedure reported by Jensen et al.30 XRD diffractograms were recorded for a mixture of TiO2 and CaF2 (50%, w/w) and the areas of the 100% peaks of rutile (110) and CaF2 (220) were determined. By comparing the ratio between the areas of (110) and (220) peaks to the ratio obtained by using the pure phase (0.90 for rutile), the amount of crystalline and amorphous phases present in the samples was determined.
SEM images were obtained using an ESEM microscope (Philips, XL30) operating at 25 kV. A thin layer of gold was evaporated on the catalyst samples, previously sprayed on the stab and dried at room temperature. TEM images were obtained using a microscope (FEI BIOTWIN G2) operating at 120 kV. BET specific surface areas were measured by the single-point method using a Micromeritics Flow Sorb 2300 apparatus. Before the measurement, the samples were dried for 1 h at 100 °C, for 2 h at 150 °C and degassed for 0.5 h at 150 °C. Thermogravimetric analyses (TGA) were performed by using Shimadzu equipment (model TG60H). The heating rate was 10 °C min−1 in static air and the powder amount, put in open Pt crucible, was 12 mg for all the samples.
2.2 Photoreactivity set up and procedure
A cylindrical Pyrex batch photoreactor with an immersed lamp, containing 0.5 L of aqueous suspension, was used to perform the reactivity experiments. The photoreactor was provided with ports in its upper section for the inlet and outlet of oxygen and for sampling. A magnetic stirrer guaranteed a satisfactory suspension of the photocatalyst and the homogeneity of the reaction mixture. A 125 W medium pressure Hg lamp (Helios Italquartz, Italy), axially positioned within the photoreactor, was cooled by water circulating through a Pyrex thimble; the temperature of the suspension was about 300 K. The radiation energy impinging on the suspension had an average value of 8 mW cm−2. It was measured at 360 nm by using a radiometer UVX Digital. The initial concentration of all the used substrates was 1 mM. Since the surface area and the agglomerate size of HPRT and SA were different, the photoreactivity runs were carried out at equal irradiation conditions of the suspension, i.e. at equal flow of photons absorbed by the suspensions. For each catalyst the amount chosen for the photocatalytic runs was that for which the resulting suspension was able to absorb ca. 90% of the impinging radiation. This choice also guaranteed that all the catalyst particles were irradiated. Measurements of the transmitted light in the presence of the suspension were carried out by using the radiometer described above. Therefore, an amount of 0.2 g L−1 was used for HPRT and 0.5 g L−1 for the commercial SA catalyst. Before switching on the lamp, oxygen was bubbled into the suspension for 30 min at room temperature to reach the thermodynamic equilibrium. In order to determine the amount of substrate adsorbed in the dark, the concentration values of substrates were routinely determined in the solution before the addition of the catalyst and in the suspension before starting the irradiation. During the run, samples of reacting suspension were withdrawn at fixed time intervals; they were immediately filtered through a 0.45 μm hydrophilic membrane (HA, Millipore) before being analysed. The quantitative determination and identification of the species present in the reacting suspension was performed by means of a Beckman Coulter HPLC (System Gold 126 Solvent Module and 168 Diode Array Detector), equipped with a Phenomenex Synergi 4 μm Hydro-RP 80A column at 298 K, using Sigma Aldrich standards. Retention times and UV spectra of the compounds were compared with those of standards. The eluent consisted of: 55% methanol and 45% 1 mM trifluoroacetic acid aqueous solution. The flow rate was 0.4 cm3 min−1. TOC analyses were carried out by using a 5000 A Shimadzu TOC analyser. All the used chemicals were purchased from Sigma Aldrich with a purity > 98.0%.3 Results and discussion
3.1 Characterization of the photocatalysts
Fig. 1 shows XRD patterns of HPRT and the commercial TiO2 photocatalyst. The peaks assignable to rutile are those at 2θ = 27.5°, 36.5°, 41°, 54.1° and 56.5°. It may be noted that the peaks of commercial rutile are well defined while those of the HPRT sample are broad, indicating a low degree of crystallinity. This feature is confirmed by the value of amorphous phase percentage present in the HPRT sample: 87.2% (ca. 0% for SA sample).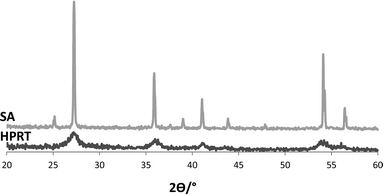 | ||
| Fig. 1 XRD patterns of commercial TiO2 (SA) and HPRT. | ||
Owing to the fact that both the TiCl4 hydrolysis and the subsequent condensation steps were carried out in a strongly acidic solution,27,31,32 only nuclei of the thermodynamically stable rutile phase were formed even if the process was carried out at room temperature. The rutile crystals needed about 6 days for their growth since the temperature was low and no additional acids (such as HNO3) were used.
The size value of primary crystallites (calculated using the Scherrer equation) was quite low for HPRT (6.8 nm) with respect to that of SA (52 nm). The size of the crystallite aggregates of HPRT was much higher than SA, as estimated from SEM and TEM observations (ca. 36 toward 240 nm) (see Fig. S1 and S2). BET specific surface areas of HPRT and SA samples were 105 and 2 m2 g−1, respectively.
TGA curves are shown in Fig. 2. For HPRT, the TGA curve shows a total weight loss of 12% due to evaporation of the physically adsorbed water (ca. 7%) and phase transformation from amorphous to crystalline. However, no weight loss was observed in the presence of SA. Even if the specific surface areas of samples are very different and therefore the TGA data might be normalized to SSA values, the curves clearly show that HPRT has a hydrophilic surface and it is mainly amorphous, while SA has a hydrophobic surface and completely crystalline structure.
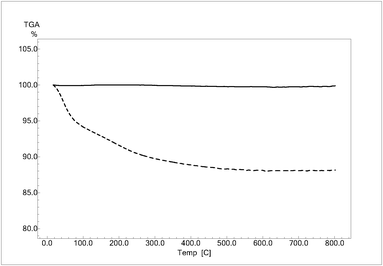 | ||
| Fig. 2 TGA curves in air of HPRT (dotted lines) and SA (continuous lines). | ||
3.2 Photoreactivity
No oxidation of alcohols was observed in the absence of irradiation, catalyst or oxygen for runs carried out under the same experimental conditions used for the photocatalytic ones. Near-UV radiation, catalyst and oxygen were all needed for the occurrence of the process.In order to investigate the effect of methoxy and hydroxy substituent groups in different single or multiple positions (ortho, meta or para) on the photooxidation process of benzyl alcohol, aqueous suspensions of benzyl alcohol were firstly investigated.
In the course of the photocatalytic runs, the main products of aromatic alcohols oxidation were the corresponding aldehydes and CO2. Table 1 reports the structural formulas of the aldehydes together with those of the corresponding substrates. A thorough investigation of other intermediates produced in the course of the photocatalytic degradation was beyond the aim of this paper. Small amounts or traces of hydroxylated aromatics, corresponding benzoic acids and open-ring products were also found.26 The amounts of these last products depended on the used photocatalyst; in the presence of HPRT, only traces of them were found, while for the commercial sample, the amounts were higher. These products obviously arise from the subsequent oxidation of produced aldehyde, this pathway being more relevant for the less selective and therefore more oxidizing sample.





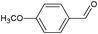
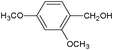
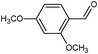

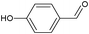


For a representative run carried out by using the HPRT sample, Fig. 3 reports the concentration values of 4-MBA, 4-methoxybenzyl aldehyde (4-MBAD) and CO2versus irradiation time. The values of the 4-MBA conversion and selectivity to aldehyde are also reported. The values of CO2 concentration have been divided by the carbon atom number of 4-MBA molecule in order to be comparable with the other values reported in the Figure. This normalization of CO2 data has been used for all the tested substrates.
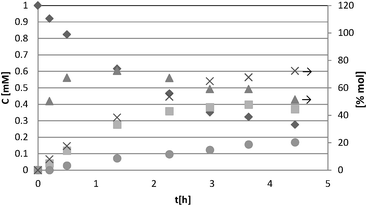 | ||
Fig. 3 Experimental results of photocatalytic oxidation of 4-MBA using HPRT (0.2 g L−1) catalyst. The CO2 concentration values were divided by 8 for normalization purposes. Conversion and selectivity values are quoted on the right ordinate axis. Symbols: concentration of: (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) 4-MBA, (■) 4-MBAD, and (●) CO2/8; (x) conversion, (▾) selectivity. ) 4-MBA, (■) 4-MBAD, and (●) CO2/8; (x) conversion, (▾) selectivity. | ||
From the data reported in Fig. 3 it may be noted that the slope of CO2 concentration data at zero time is different from zero, thus indicating that the initial CO2 is not produced through subsequent oxidation steps of the initially produced 4-MBAD released to the liquid phase. The photoreactivity runs carried out with all the other substrates showed the same feature, i.e. the rates of formation of aldehydes and CO2 had values different from zero once the irradiation was turned on. These results confirm that the aromatic alcohol oxidation proceeds through two parallel pathways, both effective since the start of irradiation: (i) partial oxidation of the aromatic alcohol to the corresponding aldehyde, which is released to the liquid phase and subsequently may compete with the alcohol for further oxidation until mineralization; (ii) complete oxidation of alcohol to CO2 and H2O through intermediates which do not desorb from the catalyst surface.2,3
The 4-MBAD selectivity values reached an almost constant value in the 4-MBA conversion range of 20–80%. Until 20% conversion, the selectivity is low probably because of the occurrence of transient phenomena on the catalyst surface;31 indeed, it is useful to report that the TOC and 4-MBAD quantitative determinations at low 4-MBA conversion may be strongly affected by experimental error. For conversions higher than 40% the selectivity decreases along with the 4-MBAD concentration in the liquid phase (see Fig. 3). In this condition the produced aldehyde molecules favourably compete with the remaining alcohol molecules for adsorption and (photo)oxidation onto the surface sites. Aldehyde concentration in the liquid phase strongly decreased especially after 70% conversion. On the basis of this finding the comparison of catalyst performances has been done by determining the selectivity and the time needed for achieving a substrate conversion of 50% and 70%.
Table 2 reports photocatalytic oxidation results of BA, 2-MBA, 3-MBA, 4-MBA, 2,4 DMBA, 4-HBA, and 4-H-3-MBA in the presence of HPRT and SA TiO2 photocatalysts. The initial degradation rate, −r0, has been determined by the following equation:
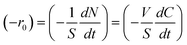 | (1) |
| Substrate | Dark ads. (%) | (−r0) × 103 (mM m h−1) | S X=0.5 (%) | S X=0.7 (%) | t X=0.5 (h) | t X=0.7 (h) | C balanceX=0.7 (%) | MinX=0.7 (%) |
|---|---|---|---|---|---|---|---|---|
| HPRT catalyst | ||||||||
| BA | 5.59 | 0.00344 | 22 | 13 | 10 | 14.5 | 91 | 78 |
| 2-MBA | 13.8 | 0.01723 | 56.5 | 50 | 2.7 | 3.65 | 99 | 49 |
| 3-MBA | 5.98 | 0.00738 | 35 | 28 | 5.0 | 9.0 | 89 | 61 |
| 4-MBA | 2.46 | 0.01662 | 70 | 59 | 2.1 | 4.0 | 90 | 31 |
| 2,4-DMBA | 4.20 | 0.02262 | 60 | 58 | 0.9 | 1.25 | 86 | 28 |
| 4-HBA | 10.2 | 0.00464 | 13 | 8.0 | 7.2 | 11 | 99 | 91 |
| 4-H-3-MBA | 12.9 | 0.00757 | 1.8 | 1.0 | 4.0 | 5.8 | 85 | 84 |
| Sigma-Aldrich catalyst | ||||||||
| BA | 2.13 | 0.113 | 8.5 | 5.0 | 6.3 | 10.1 | 36 | 31 |
| 2-MBA | 6.25 | 0.194 | 16 | 14 | 3.0 | 4.0 | 68 | 54 |
| 3-MBA | 5.74 | 0.168 | 10 | 6.0 | 4.6 | 8.0 | 68 | 62 |
| 4-MBA | 7.22 | 0.191 | 32 | 25 | 3.8 | 7.0 | 83 | 58 |
| 2,4-DMBA | 6.66 | 0.204 | 20 | 20 | 2.2 | 3.1 | 58 | 38 |
| 4-HBA | 5.40 | 0.0819 | 8.5 | 5.0 | 8.3 | 12.5 | 67 | 62 |
| 4-H-3-MBA | 3.26 | 0.114 | 3.5 | 2.2 | 4.5 | 6.6 | 57 | 55 |
The carbon mass balance, verified by taking into account only the unreacted alcohol, the produced aldehyde and CO2, was quite satisfied for the HPRT sample while for the SA one it was greatly lacking. It may be noted from the data of Table 2 that the failure on carbon mass balance increases with the decrease of the selectivity, this parameter showing the lowest values for substrates oxidized in the presence of SA catalyst.
It may be noted that with respect to the methoxy derivates, benzyl alcohol shows the lowest photocatalytic reaction rate and aldehyde selectivity while the highest selectivity was found for 4-MBA photooxidation. In the presence of methoxy group, both reaction rate and selectivity increase. The inductive and delocalization effects determined by the methoxy group on the aromatic ring hinder oxidant attacks26,27 which would cause substrate mineralization. Both HPRT and SA catalysts show the same behaviour with respect to reaction rate and aldehyde selectivity; the values of those parameters decrease in the order: 4-MBA > 2-MBA > 3-MBA > BA. The values obtained with 4-MBA and 2-MBA, however, are much closer than the other ones. This is probably due to the fact that, because the electron donor groups are ortho–para orienting, the methoxy group in the para or ortho position is able to activate the benzyl group thus favouring the alcohol-to-aldehyde transformation. In other words, two substituent groups, such as CH3O– and –CH2OH, must be mutually in the ortho or para position in order for the alcoholic group to be selectively oxidized to the aldehydic one. For this reason the donor group in the meta position is not able to affect very much the partial oxidation of substrates to aldehyde. By comparing the ortho substituent group with the para one, it may be noted that the ortho substituted substrate showed less activity and selectivity, due to hindering steric effects.
In the presence of two methoxy groups, the overall oxidation rate greatly increases indicating that the presence of more electron donor groups on the aromatic ring activates the benzyl group and destabilises the ring. In these conditions, it is easier both to oxidize the alcoholic group to aldehyde and to break the ring. The overall oxidation rate of aromatic alcohol increases together with that of aldehyde production but the selectivity decreases. The results indicate that 2,4-DMBA shows a selectivity lesser than that of 4-MBA, but in all cases higher than those of other substrates.
As to the effect of the hydroxyl group, the data of Table 2 indicate that even though both methoxy and hydroxyl are electron donor groups, the methoxy group more positively affects selectivity and activity than the hydroxyl group. By using both catalysts, it was shown that the selectivity values for 4-HBA partial oxidation are similar to those obtained with BA. This behaviour is probably due to the fact that 4-HBA could interact with the TiO2 surface by its hydroxyl group and, as a consequence, its delocalisation effect should strongly decrease. In the presence of both groups (–OH and –OCH3), high activity and low selectivity were observed. Probably the contemporary presence of these groups does not activate the benzyl group but on the contrary it destabilizes the aromatic ring.
The results obtained by the reactivity experiments indicate that the partial oxidation of aromatic substrates requires aromatic molecule interaction with the catalyst surface through the alcohol group (the aromatic ring not being involved in the adsorption) while total oxidation requires the interaction of the aromatic ring with the surface. The enhancement of mineralization with respect to partial oxidation when using crystalline rutile indicates that the generation of partial oxidation products is linked to the presence of amorphous titania on the rutile surface. The photocatalytic active sites for partial photo-oxidation of aromatic compounds, probably, are low coordinated Ti cations, characteristic of amorphous titania, where the organic molecules can adsorb with their aromatic ring plane perpendicular to the catalyst surface. The ordered structure of the rutile crystal planes should favor organic molecules adsorption with their aromatic plane parallel to the catalyst surface facilitating total oxidation.
The main oxidation products detected from the start of irradiation were the corresponding aldehydes and CO2, this last finding confirming that the mineralization of adsorbed alcohol molecules occurs by means of subsequent oxidative steps producing species which do not desorb from the photocatalyst surface to the solution. On this basis, the mineralization process should be favoured by using a catalyst which has a hydrophobic surface.21,22,24 It has been reported21,24 that low crystalline catalysts, such as the HPRT sample used in the present work, have hydrophilic surface while very crystalline catalysts, such as the SA one, have hydrophobic surfaces. Moreover, the aromatic ring of substrates could be easily adsorbed onto the crystalline rutile surface due to the fact that both of them have hydrophobic character. The hydrophilic/hydrophobic character is linked to the density of hydroxyl groups on the titania surface; this density decreases by increasing the preparation temperature of catalyst.24 The fact that HPRT catalyst was prepared at room temperature while commercial SA was obtained at high temperature could explain why HPRT is much more selective than SA for aldehyde production.
In the case of 4-H-3-MBA oxidation, it is interesting to note that HPRT showed higher mineralization percentage and less selectivity to aldehyde than SA, a behaviour different from that shown by the other aromatic alcohols. Up to now it has been reported that the selectivity values for aromatic alcohol oxidation in water are high by using less crystalline, amorphous TiO2 catalysts. In the present case, a very crystalline photocatalyst as SA is suitable for 4-H-3-MBA partial oxidation. Indeed, the selectivity values depend on the competition between the two oxidation pathways (partial or total); surface physico-chemical and structural properties of the photocatalysts are the most important factors influencing that competition, although other parameters like structure and chemical properties of the starting aromatic molecule must also be taken into account. Work is in progress in order to understand this unexpected result.
4 Conclusions
The effect of the substituent group positions on the aromatic alcohol reactivity and aldehyde selectivity was investigated by using some methoxy and hydroxy substituted benzyl alcohol derivatives in aqueous suspensions of titania under UV irradiation. The photocatalysts used for these reactions were a rutile home-prepared in mild conditions and a commercial rutile. It was observed that the home prepared catalyst was much more selective and active for aldehyde production. Moreover, para and ortho substituted aromatic alcohols were much more selectively transformed to aldehydes than the meta substituted alcohol, both catalysts exhibiting this behaviour. In the presence of more substituent groups, the reaction rate increased and selectivities decreased. The results also show that the reaction rate and selectivities depend on the kind and position of the substituent groups and on the structural properties of the catalysts.Acknowledgements
SY would like to thank Dr Levent Özcan (University of Afyon Kocatepe) for help given in TGA analysis.References
- A. Fujishima, K. Hashimoto and T. Watanabe, TiO2 Photocatalysis, Fundamentals and Applications, BKC Inc., Tokyo, 1999 Search PubMed.
- G. Palmisano, E. García López, G. Marcì, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46, 7074 RSC.
- L. Palmisano, V. Augugliaro, M. Bellardita, A. Di Paola, E. García López, V. Loddo, G. Marcì, G. Palmisano and S. Yurdakal, ChemSusChem, 2011, 4, 1431 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- V. Augugliaro, M. Litter, L. Palmisano and J. Soria, J. Photochem. Photobiol., C, 2006, 7, 127 CrossRef CAS.
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425 RSC.
- H. Yoshida, C. Murata and T. Hattori, Chem. Commun., 1999, 1551 RSC.
- K. V. S. Rao and M. Subrahmanyam, Photochem. Photobiol. Sci., 2002, 1, 597 Search PubMed.
- A. Maldotti, R. Amadelli, L. Samiolo, A. Molinari, A. Penoni, S. Tollari and S. Cenini, Chem. Commun., 2005, 1749 RSC.
- M. A. Gonzalez, S. G. Howell and S. K. Sikdar, J. Catal., 1999, 183, 159 CrossRef CAS.
- G. Palmisano, M. Addamo, V. Augugliaro, T. Caronna, E. García-López, V. Loddo and L. Palmisano, Chem. Commun., 2006, 1012 RSC.
- F. Soana, M. Sturini, L. Cermenati and A. Albini, J. Chem. Soc., Perkin Trans. 2, 2000, 699 RSC.
- T. Caronna, C. Gambarotti, L. Palmisano, C. Punta and F. Recupero, J. Photochem. Photobiol., A, 2005, 171, 237 CrossRef CAS.
- B. Ohtani, S. Tsuru, S.-I. Nishimoto, T. Kagiya and K. Izawa, J. Org. Chem., 1990, 55, 5551 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., 2002, 114, 4720 CrossRef CAS; K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538 CrossRef.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636 CrossRef CAS.
- O. S. Mohamed, A. E. M. Gaber and A. A. Abdel-Wahab, J. Photochem. Photobiol., A, 2002, 148, 205 CrossRef CAS.
- S. Higashimoto, N. Kitao, N. Yohida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279 CrossRef CAS.
- U. R. Pillai and E. Sahle-Demessie, J. Catal., 2002, 211, 434 CAS.
- G. Palmisano, S. Yurdakal, V. Augugliaro, V. Loddo and L. Palmisano, Adv. Synth. Catal., 2007, 349, 964 CrossRef CAS.
- V. Augugliaro, H. Kisch, V. Loddo, M. J. López-Muñoz, C. Márquez-Álvarez, G. Palmisano, L. Palmisano, F. Parrino and S. Yurdakal, Appl. Catal., A, 2008, 349, 182 CrossRef CAS.
- V. Augugliaro, H. Kisch, V. Loddo, M. J. López-Muñoz, C. Márquez-Álvarez, G. Palmisano, L. Palmisano, F. Parrino and S. Yurdakal, Appl. Catal., A, 2008, 349, 189 CrossRef CAS.
- M. Addamo, V. Augugliaro, M. Bellardita, A. Di Paola, V. Loddo, G. Palmisano, L. Palmisano and S. Yurdakal, Catal. Lett., 2008, 126, 58 CrossRef CAS.
- V. Augugliaro, V. Loddo, M. J. López-Muñoz, C. Márquez-Álvarez, G. Palmisano, L. Palmisano and S. Yurdakal, Photochem. Photobiol. Sci., 2009, 8, 663 CAS.
- S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568 CrossRef CAS.
- V. Augugliaro, T. Caronna, V. Loddo, G. Marcì, G. Palmisano, L. Palmisano and S. Yurdakal, Chem.–Eur. J., 2008, 14, 4640 CrossRef CAS.
- S. Yurdakal, G. Palmisano, V. Loddo, O. Alagöz, V. Augugliaro and L. Palmisano, Green Chem., 2009, 11, 510 RSC.
- V. Loddo, S. Yurdakal, G. Palmisano, G. E. Imoberdorf, H. A. Irazoqui, O. M. Alfano, V. Augugliaro, H. Berber and L. Palmisano, Int. J. Chem. React. Eng., 2007, 5, A57 Search PubMed.
- S. Yurdakal, V. Loddo, G. Palmisano, V. Augugliaro, H. Berber and L. Palmisano, Ind. Eng. Chem. Res., 2010, 49, 6699 CrossRef CAS.
- H. Jensen, K. D. Joensen, J. E. Jørgensen, J. S. Pedersen and E. G. Søgaard, J. Nanopart. Res., 2004, 6, 519 CrossRef CAS.
- S. Yang, Y. Liu, Y. Guo, J. Zhao, H. Xu and Z. Wang, Mater. Chem. Phys., 2003, 77, 501 CrossRef CAS.
- S. Yurdakal, V. Loddo, G. Palmisano, V. Augugliaro and L. Palmisano, Catal. Today, 2009, 143, 189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM images, XRD diffractograms. See DOI: 10.1039/c2ra20960h |
| This journal is © The Royal Society of Chemistry 2012 |