Nafion®-stabilised Pt/C electrocatalysts with efficient catalyst layer ionomer distribution for proton exchange membrane fuel cells†
Oliver J.
Curnick
a,
Bruno G.
Pollet
b and
Paula M.
Mendes
*a
aSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: P.M.Mendes@bham.ac.uk
bHySA Systems Competence Centre, SAIMAC, University of the Western Cape, Bellville, Cape Town 7505, South Africa
First published on 27th June 2012
Abstract
We report a facile method for the preparation of proton exchange membrane fuel cell (PEMFC) electrocatalysts from Nafion®-stabilised colloidal Pt nanoparticles (Nafion®-Pt/C), offering synthetically-directed formation of the Pt-ionomer interface and providing unprecedented control over the morphology of Pt particles on the carbon support. Electrochemical characterisation of the catalysts in aqueous acidic electrolytes using Rotating Disc Electrode (RDE) techniques revealed that Nafion®-Pt/C catalysts possessed similar specific activity and mass activity towards the oxygen reduction reaction (ORR) as commercial Pt/C catalysts, whilst requiring lower overall ionomer loadings. Combined with the near-100% utilisation measured for the Nafion®-Pt/C catalysts, this implies that Pt nanoparticles synthesised with Nafion® as a stabiliser can be ‘tuned’ to have simultaneous access to the reactant gas, the electron conducting carbon support and the proton conducting polymer electrolyte in the catalyst layer, thereby optimising the triple-phase reaction zone. By taking advantage of this bottom-up approach, which allows nanoscale control of the Nafion®–Pt–carbon interface, new opportunities exist to lower the Pt loading and the cost of the fuel cell.
Introduction
Proton exchange membrane fuel cells (PEMFCs) have been the focus of intensive research in the last decade due to their potential for environmentally friendly and highly efficient electric power generation.1 Because of their distinctive properties, PEMFCs have been considered for a wide variety of power applications ranging from portable and stationary power supplies to transportation.2,3 However, some of the most critical barriers to their commercialisation have been the intrinsic high cost and poor long-term durability of Pt-based catalysts which are currently required for practical fuel cell systems.4 More durable catalysts with lower Pt content are therefore required in order to achieve commercial viability for PEMFCs in mass-market applications.Optimisation of catalyst utilisation (the fraction of catalyst particles within the electrode which are electrocatalytically active) provides a route for lowering Pt loading, which has received relatively little attention to date compared with, for example, the production of bimetallic catalysts with inherently higher catalytic activity,5 or the use of novel electrocatalyst support materials.6,7 The extent of catalyst utilisation is closely dependent upon the structure-related properties of the PEMFC catalyst layers, which form the electrochemically-active components of the electrodes.8,9 The catalyst layers typically consist of Pt nanoparticles supported on electrically conductive carbon black particles that are partially embedded in a proton conducting polymeric ionomer, such as Nafion®.10 The triple-phase boundary (TPB) concept holds that the catalytic reactions—anodic oxidation of hydrogen (Hydrogen Oxidation Reaction – HOR) and cathodic reduction of oxygen (Oxygen Reduction Reaction – ORR)—can only occur at confined spatial regions where proton-conducting electrolyte (Nafion® or water), reactant gas, and electrically connected catalyst particles are in contact.9,11–13 Thus, a catalyst layer with an extensive, high-density distribution of triple-phase boundaries is important for the development of high Pt utilisation catalysts.
On the basis of the current state of PEMFC technology, there is still an urgent need to engineer optimum triple-phase boundary-supported fuel cell catalysts in order to maximise Pt utilisation and lower the overall cost of PEMFC systems.
In a standard catalyst layer preparation, it has become commonplace to add a Nafion® ionomer dispersion to the catalyst ink containing the Pt/C catalyst before it is applied to the substrate (PEM or GDL). Several studies have shown that the added Nafion® creates a proton-conducting network throughout the macroporous and mesoporous structure formed by the agglomeration of carbon particles upon drying,9 but that it does not penetrate into the micropores (<2 nm diameter) within the carbon support.14 This implies that the Pt nanoparticles residing within the carbon micropores are isolated from the proton-conducting network and do not contribute to the catalytic reactions, leading to significant losses in active Pt surface area within the catalyst layer. Furthermore, experimental and theoretical investigations have shown that it is difficult even to approach complete ionomer coverage of Pt particles within mesopores when using the standard chemical preparation route. Attempting to increase coverage by addition of ionomer beyond about 33 wt% Nafion® results in reduced performance as a result of electrical isolation of catalyst agglomerates and blockage of gas diffusion pathways by excessive ionomer. In order to overcome these limitations, we have investigated the synthesis of Nafion®-coated Pt nanoparticles wherein the degree of coating and ionomer connectivity may be tuned to achieve complete catalyst utilisation at lower overall ionomer loading. We propose that by partially-functionalising Pt nanoparticles with Nafion® prior to addition of the carbon support, they will not only be accessible to the ionomer network but also on the bare regions to the reactant gas and the electron conducting carbon support in the catalyst layer, forming an effective triple-phase reaction zone at the nanoscale. Previously, we15,16 and others17,18 have shown that ionomer-stabilised Pt catalysts offer enhanced durability with respect to Pt/C catalysts prepared via standard methods.15–18 However, the effect of this preparation route on catalyst utilisation and ORR activity has not been thoroughly investigated to date.
Experimental
Synthesis of Nafion®-stabilised colloidal Pt nanoparticles
The crucial step in the production of the Nafion®-Pt/C catalyst is the synthesis of Nafion®-stabilised colloidal Pt nanoparticles, which are then purified to remove excess Nafion® and supported on carbon black. All glassware was cleaned with aqua regia before use, and rinsed using UHQ water (Millipore). In a typical synthesis, Nafion® dispersion (10 wt% in water and lower aliphatic alcohols, EW 1100, Ion Power) was added to a 5 ml aliquot of 20 mM H2PtCl6·6H2O (>99.9% Sigma) solution to achieve a Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nafion® mass ratio of 1:30. The Pt(IV) concentration was then adjusted to 2.1 mM by addition of UHQ water. After stirring at 600 rpm for 30 mins, a 500 μl aliquot of 87 mM NaBH4 (ACS Reagent, Sigma) solution was then infused through a stainless steel needle at 48.5 ml min−1 using a syringe pump.
:Nafion® mass ratio of 1:30. The Pt(IV) concentration was then adjusted to 2.1 mM by addition of UHQ water. After stirring at 600 rpm for 30 mins, a 500 μl aliquot of 87 mM NaBH4 (ACS Reagent, Sigma) solution was then infused through a stainless steel needle at 48.5 ml min−1 using a syringe pump.
The as-synthesised colloidal Pt contains an excess of Nafion®, such that a catalyst layer derived from this product would contain over 90% Nafion® by volume, along with various impurities (including Cl−). The performance of such a catalyst layer would be very poor, owing to the loss of pore space required for gas transport and water removal, and poisoning of the catalyst by impurities. Excess ionomer was removed by 1:1 v/v dilution of the as-synthesised colloidal Pt with acetone, followed by centrifugation at 25000 × g for 2 h, whereupon complete precipitation of the nanoparticles occurred. The supernatant containing excess Nafion® and soluble impurities from the synthesis was discarded, and the precipitate re-dispersed in 1:1 v/v water/acetone. The Nafion® content was determined by thermogravimetric analysis (TGA, Netzsch F1 Iris) performed in air between 25 °C and 800 °C, with a 50 °C min−1 temperature ramp.
Preparation of Nafion®-Pt/C catalysts
An aliquot of Vulcan XC-72R carbon black (Cabot) dispersed in 20/80 v/v isopropanol (Sigma)/water was added to the colloidal Pt under sonication for 30 min (40 kHz, bath) to achieve the desired Pt/C ratio. Catalysts containing both 20 wt% and 50 wt% Pt were prepared by adjusting the quantity of carbon dispersion added.Transmission electron microscopy (TEM) imaging was used to confirm adherence of Pt nanoparticles to the carbon support.
In order to determine the optimal Nafion® content for the new catalyst, a series of inks were formulated from the as-prepared Nafion®-Pt/C catalysts containing several different quantities of Nafion® (10, 15, 20, 25, 30, 35 wt% in the dry catalyst layer) by addition of the appropriate quantities of Nafion® dispersion.
Electrochemical characterisation
Electrochemical characterisation was carried out in a 3-electrode electrochemical cell using an Autolab PGSTAT302N potentiostat with a home-made reversible hydrogen reference electrode (RHE) and a Pt gauze counter electrode in 0.1 M perchloric acid (TraceSelect, Sigma). All potentials in this work are referred against the RHE at 298 K. Working electrodes were prepared by drop-casting a known volume of the Nafion®-Pt/C catalyst ink onto a glassy carbon disc (5 mm diameter, Pine Instruments) pre-coated with 1 μl ethylene glycol (Sigma) under rotation at 100 rpm, to achieve a Pt loading of 20 μg Pt cm−2, before drying in a vacuum oven overnight at 313 K. For comparison, working electrodes were also prepared at the same Pt loading using two commercial Pt/C catalysts (E-Tek HP 50 wt% Pt/C; TKK TEC10E50E 46 wt% Pt/C) dispersed in 80/20 v/v water/isopropanol. Nafion® dispersion was added to aliquots of the commercial inks to produce two series of inks with a range of Nafion® loadings (20, 27, 33, 40%) in order to verify the optimal 33 wt% Nafion® reported by various research groups for this catalyst. Catalysed working electrodes were mounted in a Rotating Disc Electrode (RDE) assembly (Pine Instruments) for testing.Specific electrochemical surface area (ECSA) was determined from the charge due to desorption of underpotentially-deposited hydrogen (QHupd) measured from cyclic voltammograms recorded at 25 mV s−1 between +0.05 V and +1.0 V vs. RHE in N2-purged electrolyte, after subtraction of capacitive currents and assuming a charge density of 210 μC cm−2 for the desorption of a monolayer of adsorbed hydrogen.
Measurements of the ORR kinetics were carried out after purging the electrolyte solution with O2 (ultra-high purity, grade 5.0, BOC) for at least 30 min. Potential was swept anodically at 25 mV s−1 from +0.05 V to +1.10 V vs. RHE at various RDE rotation speeds (400, 505, 660, 933, 1,286 and 2,000 rpm). Background currents were removed by subtracting data recorded over the same potential range in N2-purged electrolyte, and corrections were applied to compensate for ohmic resistance, measured by electrochemical impedance spectroscopy at 10 kHz.
TEM characterisation
Size distributions for the Pt particles were determined from TEM micrographs recorded using a JEOL 1200ex instrument operating at 80 kV, by measuring at least 100 particles. Physical surface areas were estimated from these size distributions by assuming spherical particles.Results and discussion
Synthesis of Nafion®-stabilised colloidal Pt nanoparticles
In a typical synthesis, the clear, yellow chloroplatinic acid/Nafion® mixture turned dark brown immediately upon addition of sodium borohydride, with concomitant evolution of hydrogen gas. The colour darkened over the course of a few minutes, after which no further changes were observed. The formation of metallic nanoparticles is confirmed by the UV spectra, which showed the disappearance of the peak at 264 nm due to the PtCl62− anion, and the evolution of a characteristically featureless spectrum in the range 250–750 nm.Control of Pt particle size and morphology
The Nafion®:Pt mass ratio was found to have a significant effect on the morphology and stability of the colloidal Pt. The characteristic TEM micrographs shown in Fig. 1 demonstrate a significant improvement in Pt particle dispersion and a reduction in the degree of agglomeration upon increasing the Nafion® content from Nafion®:Pt ratio 1:1 to 5:1 to 30:1. However, adjustment of precursor pH from an initial value of 2–3 to 5.0 using aqueous NaHCO3 solution prior to the reduction step was found to lead to extensive agglomeration of colloidal Pt particles, even at Nafion®:Pt = 30:1, as shown in Fig. 1(d).
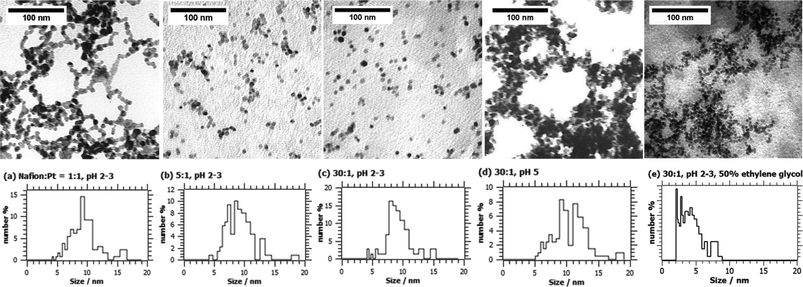 | ||
| Fig. 1 TEM images and Pt particle size distributions for as-prepared colloidal Pt synthesised at (a–c, e) pH 2-3 and (d) pH 5 with various Nafion:Pt mass ratios (a) 1:1, (b) 5:1, (c,d,e) 30:1; (e) synthesised with the addition of 50% v/v ethylene glycol to the diluant in the preparation of the H2PtCl6 precursor solution. | ||
Despite its obvious effect on particle dispersion, the size distributions in Fig. 1(a–d) reveal that the Nafion®:Pt ratio has relatively little influence on the primary Pt particle size. However, it was found that the (Sauter) mean Pt particle size could be reduced from 7.8 nm to 5.8 nm by replacing 50% of the diluent water used in the preparation of the PtCl62− precursor with ethylene glycol (EG), as shown in the particle size distribution in Fig. 1(e). Unlike a typical ‘polyol’ synthesis in which EG acts as the reducing agent, it is thought that the main effect of the additional EG in this method is to increase the viscosity of the continuous phase, thereby reducing the rate of particle growth by lowering the rate of diffusion of monatomic Pt(0) species to the surface of metal nuclei. This is supported by the fact that the product synthesised via the EG-assisted method contains a far larger proportion of small particles in the range 2–5 nm (Fig. 1(e)) than do the products synthesised without EG.
Preparation of supported catalysts
Colloidal Pt dispersions synthesised with Nafion®:Pt = 30:1 at pH 2–3 were selected for preparation of supported catalysts (Nafion®-Pt/C). For investigation of particle size effects in later electrochemical testing, supported catalysts were also prepared from colloidal Pt synthesised in the presence of EG (Nafion®-EG-Pt/C). The extent of Pt nanoparticle agglomeration on the final supported catalyst could be varied by addition of the carbon support either prior to or following purification by centrifugation. The products of these two preparation routes are designated as Nafion®-Pt/C A and Nafion-Pt/C B, respectively, and the routes are illustrated schematically in Fig. 2. The TEM micrographs in Fig. 3 show the dramatic variation in morphology between Nafion® Pt/C A and Nafion-Pt/C B catalysts, produced simply by adding the carbon support at different stages during the purification.
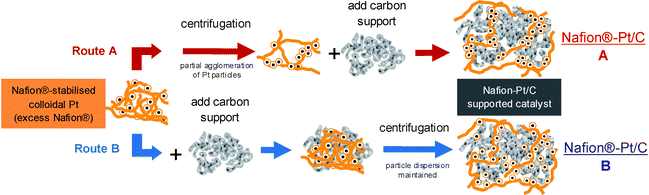 | ||
| Fig. 2 Carbon-supported catalysts can be derived from colloidal Pt via two routes: (A) the carbon support is added after removal of excess Nafion® by centrifugation; (B) the carbon support is added before centrifugation. The two routes produce markedly different Pt particle morphologies, with route A yielding loosely-agglomerated networks of Pt particles extending from the carbon support, whilst route B produces a more conventional, well-dispersed morphology. | ||
 | ||
| Fig. 3 TEM micrographs of Pt/C catalysts tested in this study. | ||
For route A, thermogravimetric determination of Nafion® content showed that successive centrifugation/re-dispersion cycles removed excess Nafion® from colloidal Pt, but that after 3 cycles there remained around 20 wt% Nafion® in the product which could not be removed by further treatment (Fig. S1 in ESI†). The precipitation of Pt particles to form loosely-agglomerated networks was observed by TEM to be concomitant with the removal of excess stabilising Nafion®. It is proposed that the residual 20 wt% Nafion® is strongly bound within these Pt agglomerates, forming a well-coordinated Pt-ionomer interface, which persists upon addition of the carbon support. Conversely in route (B), the presence of the carbon support during purification provides a high-surface area substrate onto which Pt particles are able to adsorb as they precipitate from colloidal dispersion following removal of stabilising Nafion®, thus preventing agglomeration and preserving instead the dispersion of the Nafion-Pt/C B catalysts. The morphology of the Nafion-Pt/C B catalyst resembled that of the E-Tek and TKK commercial catalysts (Fig. 3). On the other hand, the other catalyst prepared by route A, Nafion-EG-Pt/C, exhibited similar morphology to that of Nafion-Pt/C A, with agglomerated networks of Pt particles present of the carbon support.
Electrochemical characterisation
Catalysed working electrodes were prepared by pipetting a known volume of catalyst ink dispersed in 80/20 (v/v) water/IPA onto a glassy carbon RDE pre-coated with 1 μl EG, under rotation at 100 rpm followed by drying in a vacuum oven. This method was found to be reliable in yielding high-quality catalyst films which completely and uniformly covered the glassy carbon disc.Catalyst utilisation
The ECSAs of the various catalysts were measured from the hydrogen desorption charge QH calculated from cyclic voltammograms recorded at 25 mV s−1 between +0.05 V and +1.1 V in 0.1 M HClO4 according towhere LPt is the Pt loading per unit area on the electrode and Ageo is the geometric area of the elctrode. Physical surface areas APhys were measured from TEM micrographs, assuming spherical Pt particles, and catalyst utilisation was calculated according to U = ECSA/APhys
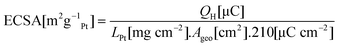
From Fig. 4(a), the optimal Nafion® content of the Nafion®-Pt/C A catalyst was found to be around 20 wt%, which is far lower than the 30–35 wt% optimum loading for conventional Pt/C catalysts reported previously and verified here for the E-Tek catalyst. Close coordination between Pt and residual Nafion® in the as-prepared Nafion®-Pt/C A catalyst is thought to provide close to 100% utilisation of Pt particles within agglomerates on the carbon support. However, the absence of free Nafion® in the as-prepared ink results in poor ionomeric connectivity between these agglomerates, such that not all are utilised. This accounts for the lower utilisation observed for Nafion-Pt/C A at low Nafion® loading in Fig. 4(a). Addition of a small amount of Nafion® to the as-prepared catalyst facilitates connection between agglomerates, resulting in a rapid increase in utilisation with increasing ionomer content, up to a maximum at about 20 wt% NFP. This optimum utilisation is obtained at a lower overall Nafion® loading indicating an enhanced percolation efficiency of the ionomer network within the Nafion®-Pt/C catalyst layer. At this optimum ionomer loading, the Nafion®-Pt/C catalyst demonstrates a two-fold increase in catalyst utilisation (95 ± 7%) compared with the conventionally-prepared E-Tek catalyst (50 ± 3%) at its corresponding optimum. Beyond this optimum ionomer content, the electrical isolation of Pt/C agglomerates by an encapsulating ionomer film has a negative impact on utilisation. The fact that 100% utilisation is measured in spite of the apparent agglomeration of Pt particles is encouraging, as it implies that the degree of Pt–Pt particle contact is sufficiently low as to have very little effect on the active surface area of the catalyst, and proves that Pt particles and agglomerates are in good electrical contact with the carbon support: it would be impossible to attain such high utilisations if significant numbers of Pt particles were electrically isolated from the support. The presence of a percolating network of Nafion® ionomer within Pt agglomerates is thought to be important for the attainment of full catalyst utilisation, whilst also limiting the extent of particle–particle contact via steric stabilisation.
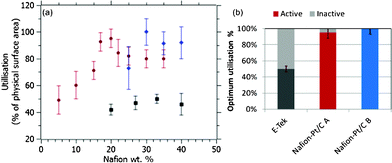 | ||
| Fig. 4 (a) Catalyst utilisation as a function of catalyst layer ionomer content for Nafion®-Pt/C A (●), Nafion®-Pt/C B (◆) and E-Tek (■) catalysts measured on working electrodes with Pt loadings of 80 μg cm−2; (b) optimum catalyst utilisations found for these three catalysts. | ||
Interestingly, the optimum Nafion® content of Nafion®-Pt/C B catalyst was found to be higher than that of Nafion®-Pt/C A. The optimum of around 30 wt% is similar to that found for the E-Tek catalyst, although the Nafion®-Pt/C B is fully utilised at this Nafion® loading, whilst the E-Tek catalyst offers only 50% utilisation. The variation in optimum Nafion® content between the Nafion®-Pt/C A and B catalysts can only be attributed to their differing morphologies, as their physical surface areas, Pt particle size distributions and Pt/C ratios are very similar. The need for a higher Nafion® loading is indicative of a less efficient ionomer percolation in the Nafion®-Pt/C B catalyst, akin to that found in a standard catalyst preparation. This finding supports the assertion that the Pt agglomerates in the Nafion®-Pt/C A catalyst contain a highly-effective internal ionomer network: it is logical that to establish connectivity between these large agglomerates should require less ionomer than for individual, well-dispersed Pt nanoparticles.
Oxygen reduction performance
Measurement of oxygen reduction kinetics was carried out using an RDE method in O2-saturated 0.1 M perchloric acid. To ensure accurate measurement of mass activity, Pt loadings of 20 μgPt cm−2 were used on working electrodes to maximise catalyst utilisation. The raw RDE data were corrected for background currents by subtracting linear scan voltammetry (LSV) scans performed at 25 mV s−1 under nitrogen purge, before correcting potentials for ohmic resistance using ΔE = IR where I is the current at each data point and R is the ohmic resistance between the working and reference electrodes determined by Electrochemical Impedance Spectroscopy (EIS) at 10 kHz.| J lim @ 2,000 rpm/mA cm−2geo | D eff/10−5 cm2 s−1 | |
|---|---|---|
| Nafion®-Pt/C A | −5.53 | 1.61 |
| Nafion®-Pt/C B | −6.29 | 2.06 |
| Nafion®-EG-Pt/C | −6.54 | 2.00 |
| E-Tek HP 50 wt (%) | −6.78 | 2.40 |
| TKK TEC10E50E | −6.79 | 2.42 |
The limiting current densities were also found to be linearly dependent on ω1/2 as predicted by the Levich equation. From the slopes of the Levich plots in Fig. 5, effective diffusion coefficients (Deff) for each electrode were calculated and are summarised in Table 1. The diffusion of O2 was found to be slightly slower for Nafion®-stabilised electrocatalysts than for both the commercial catalysts. The Nafion®-Pt/C A catalyst had the lowest effective diffusion coefficient (1.61 × 10−5 cm2 s−1), and this is thought to be due to a relatively thick layer of Nafion® encapsulating Pt agglomerates on the carbon support. This impaired mass transport may be exacerbated in gas diffusion electrodes, reducing the performance of MEAs operating at high current densities, and this will be investigated in future work.
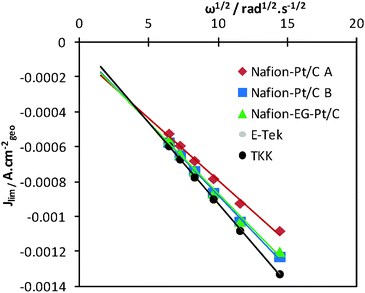 | ||
| Fig. 5 Levich plots for catalysts on test, demonstrating linear relationships between limiting current Jlim (measured at +0.3 V) and ω1/2. | ||
The background and IR-corrected RDE curves in Fig. 6(a) show variations in onset and half-wave potentials (E1/2) for ORR in the mixed kinetic/diffusion controlled region between +0.8–+1.0 V, which are due to the differences in electrode kinetics, quantified by kinetic current densities shown in the Tafel plot in Fig. 6(b). Kinetic current densities Jk in Fig. 6(b) were calculated using the Koutecky–Levich equation: Jk = J.Jlim/(Jlim − J), and at +0.9 V these correspond well with values obtained by extrapolating Koutecky–Levich plots (see ESI†). The specific activities (SA) at +0.9 V calculated using the K–L plot method and mass activities (MA) are shown in Fig. 6(c–d) and Table 2, along with Tafel slopes calculated in the range +0.88–+0.9 V.
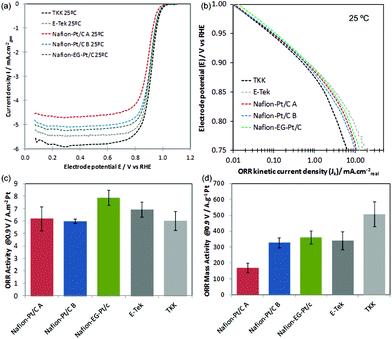 | ||
| Fig. 6 (a) IR-corrected RDE curves recorded at 1286 rpm, 25 mV s−1 in O2 saturated 0.1 M HClO4 at 298 K; (b) mass-transport corrected Tafel plots; (c) ORR specific activities and (d) ORR mass activities at +0.9 V vs. RHE. Catalysts in (c) and (d) are arranged in decreasing order of Pt particle size from left to right. | ||
| Catalyst | Particle size/nm | A Phys/m2 g−1Pt | ECSA/m2 g−1Pt | U Pt% | SA @ +0.9 V/A−1 m−2real | MA @ +0.9 V/A−1 g−1Pt | Tafel slope (b)/mV dec−1 |
|---|---|---|---|---|---|---|---|
| Nafion®-Pt/C A | 7.8 | 37 | 35 | 95 | 6.19 | 169 | −65 |
| Nafion®-Pt/C B | 5.6 | 51 | 45 | 88 | 6.00 | 328 | −58 |
| Nafion®-EG-Pt/C | 5.8 | 49 | 46 | 94 | 7.88 | 362 | −61 |
| E-Tek HP 50 wt% | 4 | 71 | 55 | 76 | 6.93 | 341 | −62 |
| TKK TEC10E50E | 2.9 | 98 | 91 | 93 | 6.02 | 507 | −62 |
The fact that Nafion®-Pt/C catalysts show similar SA to the commercial catalysts is encouraging, and shows that the reaction is not inhibited by capping Nafion® in contact with Pt surfaces. The variations in mass activity between the catalysts shown in Fig. 6(d) are in keeping with the differences in particle size and specific surface area i.e. mass activity is found to increase with decreasing particle size.
Conclusions
A method has been developed for the synthesis of stable and well-dispersed Nafion®-stabilised colloidal Pt nanoparticles with sizes in the range 3–10 nm. In preparing supported catalysts from Nafion®-stabilised colloidal Pt, the distribution of Pt particles on the carbon support can be varied between well-dispersed (Nafion®-Pt/C B) or agglomerated (Nafion®-Pt/C A) by adding the support either before or after purification, respectively. Addition of ethylene glycol to the reaction mixture allowed the synthesis of a Nafion®-Pt/C A-type catalyst with reduced Pt particle size (Nafion®-EG-Pt/C). The optimum Nafion® content for the Nafion®-Pt/C A catalyst was found to be 20% NFP, compared to ca. 33% for the commercial (E-Tek) catalyst and Nafion®-Pt/C B type catalyst. It is thought that the enhanced utilisation and lower optimum Nafion® loading are direct results of a more efficient ionomer percolation afforded by the Nafion®-Pt/C preparation route. The Nafion®-Pt/C A catalysts achieved 100% utilisation even at high Pt loadings, although in situ testing will be required to determine whether the new preparation route can improve utilisation in the much thicker catalyst layers within PEMFC electrodes, and to confirm that it does not impair mass transport and electrical conductivity. All three Nafion®-Pt/C catalysts displayed SAs comparable with E-Tek and TKK catalysts. The MA of the Nafion®-Pt/C A catalyst was considerably lower than the other catalysts by virtue of its larger Pt particle size, but the MAs of the Nafion®-Pt/C B and Nafion®-EG-Pt/C catalysts were comparable with the E-Tek catalyst, within the bounds of experimental error. Combined with the near-100% utilisation measured at lower overall ionomer content for the Nafion®-Pt/C A catalysts, this implies that Pt nanoparticles synthesised with Nafion® as a stabiliser can be ‘tuned’ to have simultaneous access to the reactant gas, the electron conducting carbon support and the proton conducting polymer electrolyte in the catalyst layer.Acknowledgements
We wish to thank the Leverhulme Trust grant number F/00 094/BD for financial support. This research was in part supported through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2).References
- W. Vielstich, A. Lamm, H. A. Gasteiger, Handbook of Fuel Cells – Fundamentals Technology and Applications; Wiley, 2003 Search PubMed.
- I. Staffell, R. Green and K. Kendall, J. Power Sources, 2008, 181, 339 CrossRef CAS.
- B. G. Pollet, I. Staffell, J. L. Shang, Electrochimica Acta, 2012, in press Search PubMed.
- H. Zhang, X. Wang, J. Zhang, J. Zhang, in PEM Fuel Cell Catalysts and Catalyst Layers; Zhang, J., Ed.; Springer, 2008, p 889 Search PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990 CAS.
- S. Shrestha, Y. Liu and W. E. Mustain, Catal. Rev. Sci. Eng., 2011, 53, 256 CAS.
- D. L. Wood, J. Chlistunoff, J. Majewski and R. L. Borup, Journal of the American Chemical Society, 2009 Search PubMed.
- K. Malek, M. Eikerling, Q. Wang, T. Navessin and Z. Liu, J. Phys. Chem. C, 2007, 111, 13627 CAS.
- E. Allahyarov, P. L. Taylor and H. Lowen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, ARTN 031805 CrossRef.
- M. Eikerling, K. Malek, J. Liu and Q. Wang, Section title: Electrochemical, Radiational, and Thermal Energy Technology, 2008, 53, 705 CAS.
- K. Ikeda, N. Nonoyama and Y. Ikogi, ECS Trans., 2010, 33, 1189 CrossRef CAS.
- X. Yu, J. Yuan and B. Sunden, J. Fuel Cell Sci. Technol., 2011, 8, 034001 CrossRef.
- A. Ignaszak, S. Ye and E. Gyenge, J. Phys. Chem. C, 2009, 113, 298 CAS.
- O. J. Curnick, P. M. Mendes and B. G. Pollet, ECS Trans., 2010, 33, 557 CrossRef CAS.
- O. J. Curnick, P. M. Mendes and B. G. Pollet, Electrochem. Commun., 2010, 12, 1017 CrossRef CAS.
- N. Cheng, S. Mu, M. Pan and P. P. Edwards, Electrochem. Commun., 2009, 11, 1610 CrossRef CAS.
- S. Yin, S. Mu, H. Lv, N. Cheng, M. Pan and Z. Fu, Appl. Catal., B, 2010, 93, 233 CrossRef CAS.
- J. J. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic and M. Arenz, Electrochim. Acta, 2008, 53, 3181 CrossRef.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stab, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354 CrossRef CAS.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA data, catalyst utilisation data measured by cyclic voltammetry, ORR kinetics data. See DOI: 10.1039/c2ra21071a |
| This journal is © The Royal Society of Chemistry 2012 |