Synthesis and spectroscopic properties of bodipy dimers with effective solid-state emission†
Lizhi
Gai
a,
Hua
Lu
*a,
Bin
Zou
a,
Guoqiao
Lai
a,
Zhen
Shen
*b and
Zhifang
Li
*a
aKey Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 310012, P. R. China. E-mail: hualu@hznu.edu.cn; zhifanglee@hznu.edu.cn; Fax: (+86) 571-2886-5135
bState Key Laboratory of Coordination Chemistry, Nanjing National Laboratory of Microstructures, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China. E-mail: zshen@nju.edu.cn; Fax: (+86) 25-8331-4502
First published on 27th July 2012
Abstract
Boron-dipyrromethenes (BODIPYs) dimers with phenyl and bulky triphenylsilylphenyl substituents were synthesized through oxidative self-coupling of the 2-position with FeCl3. Spectroscopic properties of all the dyes in various solvents and on films have been investigated. In comparison with the corresponding monomers, the dimers exhibit higher molar absorption coefficients, relative moderate fluorescent quantum yields and redshifted wavelengths. The luminescence yields of the dimers are solvent polarity dependent and decrease dramatically in acetonitrile. More intensive solid-state emission of triphenylsilylphenyl substituted BODIPY dimer is observed with a quantum yield of 9.7% relative to the phenyl substituted dimer, which could be attributed to the introduction of the bulky group that inhibits aggregation.
1. Introduction
As early as 1968, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) dyes were firstly discovered by Treibs and Kreuzer.1 Until the last two decades, BODIPY derivatives have attracted tremendous attention as fluorescence dyes due to their advantageous photophysical properties such as excellent photochemical stabilities, narrow absorption and emission bands, large molar absorption coefficients, and high fluorescence quantum yields. BODIPY dyes have widespread applications in biochemical labeling, fluorescent switches, fluorescent probes, sensitizers for solar cell and laser dyes.2However, BODIPY dyes with efficient fluorescence in the solid-state are rare, the fluorescence in the solid-state is effectively suppressed due to flat π conjugated systems in molecular structures and fluorophore aggregation is very tight in the solid phase which leads to significant quenching via π–π interactions.3 Moreover, their small Stokes shifts leads to self-quenching through energy transfer.4 In order to obtain excellent BODIPY dyes with efficient emission in the solid-state, we and other groups have attempted to introduce bulky substituents to inhibit the aggregation,5,6 the introduction of triphenylsilylphenyl group at the 2,6-position of the BODIPY core is generally pronounced for increasing the solid-state emission. More intensive solid-state emission of one side substituent (2-position) than those of both sides (2,6-position) is found i.e. 2-triphenylsilylphenyl substituted BODIPY exhibits efficient solid-state emission.6 We suppose the spectroscopic properties of this compound can be further tuned by forming a dimer at the 6-position. Recently, Ziessel and coworkers successfully prepared BODIPY dimers by using (trifluoroacetate) (PIFA)/BF3OEt2,7 then Bard's group reported that dimers and trimers could be obtained through oxidative coupling at the 2/3-position by using FeCl3 as the catalyst.8 So far, only a few papers investigated in-depth the spectroscopic properties of dimers, which exhibit interesting optical properties.7,8a,14a In this paper, we further investigated the effect of triphenylsilylphenyl substituents to increase the solid-state fluorescence of BODIPY dimers using the latter synthesized method. The detailed photophysical properties were investigated in various solvents and in the solid-state. TD-DFT was carried out to explore the potential of the arylsilyl substituents and structure-property correlations.
2. Experimental section
2.1 Materials and instrumentations
All reagents were obtained from commercial suppliers and used without further purification unless otherwise indicated. All air and moisture-sensitive reactions were carried out under nitrogen atmosphere. Dichloromethane was distilled over calcium hydride. Triethylamine was obtained by simple distillation. Ether and THF were distilled over sodium. NMR spectra were recorded on a Bruker DRX400 spectrometer and referenced to the residual proton signals of the solvent. Mass spectra were measured with a Bruker Daltonics AutoflexII TM MALDI–TOF spectrometer. HRMS were recorded on a Bruker Daltonics Apex-III spectrometer.2.2 Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C25H23BF2N2]+) m/z = 400.19, found m/z = 400.40 [M]+, 381.40 [M−F]+
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C43H37BF2N2Si]+) m/z = 658.3, found m/z = 658.4 [M]+, 639.4 [M−F]+.
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C50H44B2F4N4]+) m/z = 798.37, found m/z = 798.40 [M]+, 779.30 [M−F]+, HRMS-ESI: m/z: calcd ([C50H44B2F4N4Na]+) m/z = 821.3580; found m/z = 821.3595 [M+Na+], 779.3710, [M−F]+.
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C86H72B2F4N4Si2]+) m/z = 1314.54, found m/z = 1317.03 [M]+, 1297.69 [M−F]+, HRMS-ESI: m/z: calcd ([C86H72B2F4N4Si2Na]+) m/z = 1337.5310; found m/z = 1337.5327 [M+Na+], 1315.5426 [M+H+], 1295.5354, [M−F]+.
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C25H23BF2N2]+) m/z = 400.19, found m/z = 400.40 [M]+, 381.40 [M−F]+
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C43H37BF2N2Si]+) m/z = 658.3, found m/z = 658.4 [M]+, 639.4 [M−F]+.
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C50H44B2F4N4]+) m/z = 798.37, found m/z = 798.40 [M]+, 779.30 [M−F]+, HRMS-ESI: m/z: calcd ([C50H44B2F4N4Na]+) m/z = 821.3580; found m/z = 821.3595 [M+Na+], 779.3710, [M−F]+.
000 dm3 mol−1 cm−1); MALDI-TOF: calcd ([C86H72B2F4N4Si2]+) m/z = 1314.54, found m/z = 1317.03 [M]+, 1297.69 [M−F]+, HRMS-ESI: m/z: calcd ([C86H72B2F4N4Si2Na]+) m/z = 1337.5310; found m/z = 1337.5327 [M+Na+], 1315.5426 [M+H+], 1295.5354, [M−F]+.
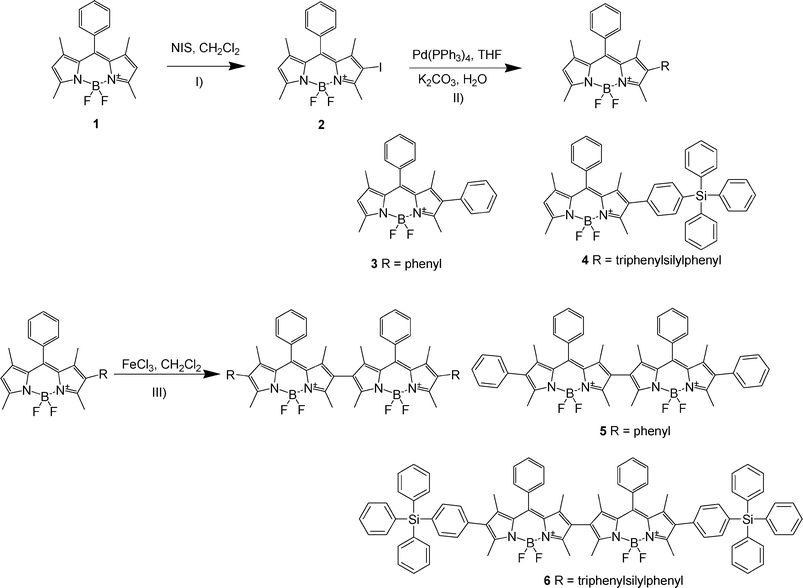 | ||
| Scheme 1 Synthesis procedure and structure of BODIPY and BODIPY dimer derivatives. I) NIS, CH2Cl2, 0–5 °C; II) Pd(PPh3)4, Ph3SiPhB(OH)2, K2CO3, THF/H2O, 80 °C; III) FeCl3, CH2Cl2, rt. | ||
2.3 Spectroscopic measurements
Uv-Vis spectra were recorded on a Shimadzu 3000 spectrophotometer. Fluorescence spectra were measured on a Hitachi F-2700 FL spectrophotometer with a xenon arc lamp as light source. Samples for absorption and emission measurements were contained in 1 cm × 1 cm quartz cuvettes. For all measurements, the temperature was kept constant at (298 ± 2 K). Dilute solution with an absorbance of less than 0.05 at the excited wavelength was used for the measurement of fluorescent quantum yields, the luminescence quantum yields in solution were measured by using rhodamine 6G with excited wavelength 488 nm (ΦF = 0.88 in ethanol) as a reference.9 The quantum yield Φ as a function solvent polarity is calculated using the following eqn (1).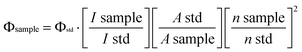 | (1) |
Where subscript sample and std denote the sample and standard, respectively, Φ is quantum yield, I is the integrated emission intensity, A stands for the absorbance, n is refractive index.
The fluorescence lifetimes of the samples were measured on a PS920 Lifespec-ps (Edinburgh) using picoseconds plused diode laser (PDL800-B) as the excitation source. The goodness of the fit of the single decays as judged by reduced chi-squared (χ2R) and autocorrelation function C(j) of the residuals was below χ2R < 1.2.
When the fluorescence decays were monoexponential, the rate constants of radiative (kr) and nonradiative (knr) deactivation were calculated from the measured fluorescence quantum yield (Φf) and fluorescence lifetime (τ) according to eqn (2) and (3):
| kr = Φf/τ | (2) |
| knr = (1−Φf)/τ | (3) |
2.4 Preparation of films
Drop casting films were prepared by evaporation of the dye (1 mM) and the PMMA containing 50 mol% dye (1 mM) in dichloromethane onto a clean silicon wafer substrates. Those films were stable and no change in emission was observed over an extended period of time. There is no difference in emission between deposits obtained by evaporating solvent and deposits obtained by stretching the solution.2.5 Computational details
The ground state structures of BODIPY were computed using the density functional theory (DFT) method with the hybrid-generalized gradient approximation (H-GGA) functional B3LYP.10 6-31G(d) basis set was assigned to the elements, which guarantees a reasonable balance of the computational cost. The absorption properties were predicted by time-dependent (TD-DFT) method with the same basis set. All of the calculations were performed with the Gaussian03 program package.113. Results and discussion
3.1 Synthesis
BODIPY 1, which was synthesized through a trifluoroacetic acid-catalyzed condensation of the corresponding benzaldehyde with two equivalents of 2,4-dimethylpyrrole, was treated with NIS giving the 2-iodo substituted BODIPY 2 with 69% yield. Phenyl and triphenylsilylphenyl BODIPY derivatives were synthesized from the iodo-BODIPY 2via reported Suzuki–Miyaura cross-coupling reaction, with phenylboronic acid and 4-triphenylsilylphenylboronic acid.12 Compounds 5 and 6 were prepared using FeCl3 as an oxidant via a one-step reaction in CH2Cl2 at 0 °C (Scheme 1). The absence of protons signal at ca 6.0 ppm and large splitting of methyl at the pyrrole ring of 5 and 6 in the 1H NMR spectra confirmed the pyrrole ring connected directly through self-oxidative coupling. High-resolution mass spectrometry (HR-MS) exhibited a molecular ion peak at 821.3595 for 5 (calcd for [M+Na]+, 821.3580) and 1337.5327 for 6 (calcd for [M+Na]+ 1337.5310), and the isotopic pattern was in accordance with the structure.3.2 Spectroscopic properties in solution
The Uv/vis absorption, fluorescent spectra and fluorescent lifetimes of 3–6 were measured in hexane, toluene, CH2Cl2, THF and CH3CN. The photophysical properties are summarized in Table 1.| Solvent | λ abs [nm] | ε max a | fwhmabs [nm] | λ em [nm] | fwhmem [nm] | Δύem−abs [nm] | Φ | τ f [ns] | κ r [10 8 s −1 ] | κ nr [10 8 s −1 ] | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a The molar absorption coefficient was measured in solution containing 1% CH2Cl2 as a co-solvent. b The optical properties of 4 in other solvents can be found in the ref. 6. | |||||||||||
| 3 | Hexane | 514 | 53000 |
26 | 534 | 30 | 20 | 0.90 | 3.49 | 2.58 | 0.29 |
| Toluene | 516 | 50000 |
26 | 538 | 32 | 22 | 0.99 | 3.44 | 2.90 | 0.03 | |
| CH2Cl2 | 514 | 46000 |
27 | 536 | 33 | 22 | 0.91 | 3.70 | 2.46 | 0.24 | |
| THF | 513 | 49000 |
27 | 536 | 34 | 23 | 0.89 | 3.59 | 2.48 | 0.31 | |
| CH3CN | 510 | 41000 |
28 | 533 | 36 | 23 | 0.76 | 3.85 | 1.97 | 0.62 | |
| 4 b | CH2Cl2 | 518 | 51000 |
29 | 538 | 34 | 20 | 0.89 | 3.42 | 2.60 | 0.32 |
| 5 | Hexane | 553 | 169000 |
33 | 582 | 33 | 29 | 0.40 | 2.69 | 1.49 | 2.23 |
| Toluene | 555 | 150000 |
35 | 584 | 34 | 29 | 0.35 | 2.55 | 1.37 | 2.55 | |
| CH2Cl2 | 552 | 151000 |
34 | 582 | 35 | 30 | 0.29 | 2.77 | 1.05 | 2.56 | |
| THF | 552 | 155000 |
35 | 582 | 34 | 30 | 0.35 | 2.70 | 1.30 | 2.41 | |
| CH3CN | 547 | 133000 |
32 | 575 | 37 | 28 | 0.01 | 0.24 | 0.42 | 41.25 | |
| 6 | Hexane | 556 | 185000 |
35 | 585 | 33 | 29 | 0.34 | 2.54 | 1.34 | 2.52 |
| Toluene | 558 | 165000 |
35 | 588 | 35 | 30 | 0.35 | 2.34 | 1.50 | 2.78 | |
| CH2Cl2 | 554 | 182000 |
35 | 585 | 36 | 31 | 0.31 | 2.64 | 1.17 | 2.61 | |
| THF | 554 | 161000 |
36 | 584 | 36 | 30 | 0.45 | 2.45 | 1.74 | 2.24 | |
| CH3CN | 549 | 186000 |
33 | 578 | 37 | 29 | 0.01 | 0.30 | 0.33 | 33.00 | |
The absorption band of the dyes 3–4 show the typical cyanine-type vibronic band which are assigned to 0–0 vibrational band of a strong S0–S1 transition. A broader, much weaker bands at the range of 350–420 nm can be attributed to the second transition.13 The dyes 5–6 show identical absorption spectra with an absorption pattern typical for BODIPY chromophores, the absorption spectra are red-shifted around 40 nm with increasing of the full width at half-maximum (fwhmabs) in comparison to the monomers 3–4 (Fig. 1). On the other hand, the increasing fwhmabs is connected to a significant increasing (>3 fold) in molar absorption coefficient. However, there is a significant difference in the spectroscopic properties of the BODIPY dimer through β–β linkage compared with the BODIPY dimer through α–α position linkage, the α–α linked BODIPY dimer shows a clearly exciton splitting and two major absorption peaks, a comparison of the extinction coefficient and the energy of the longest-wavelength absorption band with corresponding BODIPY monomer, at the same time, α–α linked BODIPY dimer has notably shifted by a Stokes shift of around 80 nm.8b,14 The absorption maxima of the compounds 3–6 are almost independent of solvent polarity with only a small variation of 6–7 nm upon going from acetonitrile to hexane, which is consistent with the general behavious of BODIPY chromophores.2b
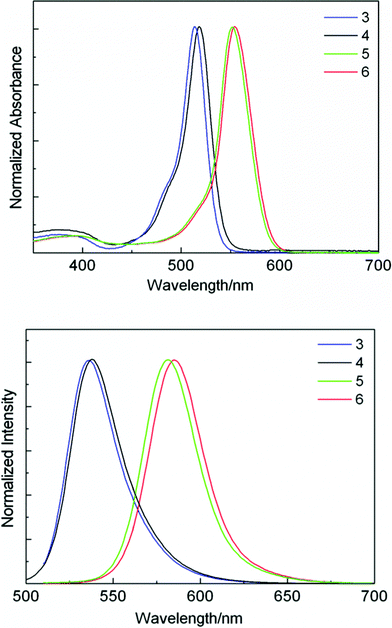 | ||
| Fig. 1 Absorption and fluorescent spectra of BODIPY derivatives 3–6 in dichloromethane. | ||
The fluorescent spectra are typical mirror-image relationship of the lowest-energy absorption spectra, the emission bands do not show any particular trend as a function of solvent polarity, these features suggest that emission occurs from the weakly polar, relaxed Franck–Condon excited state.15 Fluorescence spectra show the similar trends with a red shift around 45 nm from monomers 3–4 to dimers 5–6. The Stokes shift of 5–6 is larger than those of typical BODIPYs and the monomers 3–4, that means a geometrical rearrangement upon excition.2b,8,15 Stokes shift is independent of the solvent polarity, indicating that the permanent dipole moments do not differ between the ground state and the excited state. The absorption and fluorescent spectra of 6 with triphenylsilyl substituent are almost identical with that of 5 in different kinds of solvents except a slight red shift of 3–4 nm, indicating that the triphenylsilyl group is only barely affecting the spectroscopic properties in the solvent.
Fluorescent quantum yield of the monomer 3 is very strong and slightly dependent on the solvent polarity from toluene (0.99) to acetonitrile (0.76). For the dimers 5–6, however, a smaller value of fluorescent quantum yield and fluorescent lifetime is observed, this feature is mainly due to the enhancement of non-radiative decay, since the ground and excited state are energetically closer, the internal conversion probability is largely enhanced due to the increased vibrational coupling probability.16 On the other hand, the torsional motion of β–β linkage increase the vibrational relaxation channels, excited–state conformational changes drive faster internal conversion to the ground state, which quenches the emission.17 These trends are reflected by the non-radiative rate decay, knr = (1−Φf)/τ, a change from monomers to dimers leads to a ca 10-fold increase (Table 1). Though fluorescent quantum yield for dimers 5–6 decrease, the brightness, defined as ε × Φ, is almost unchanged compared to the monomer 3–4, without any critical solvent polarity dependent decrease of their brightness except in acetonitrile. The luminescence quantum yield and lifetime shows a trend that depends on solvent polarity and is strongly reduced in acetonitrile, consistent with a previously reported BODIPY dimer,14a probably due to solvent polarity affecting the excited-state conformation which leads to an increase the internal conversion rate, the internal conversion rate of dimers in acetonitrile is almost ca. 15-fold of other solvents (Table 1). The fluorescence decay profiles of dyes 5–6 could be described by a single-exponential fit with fluorescence lifetime in the range of 2.4–3.5 ns in all of the solvents investigated excepted for in acetonitrile (0.24–0.30 ns).
In the viscous ethylene glycol, having a similar polarity with acetonitrile, the quantum yield is ca. 0.03 and increases slightly with respect to acetonitrile, but is still much lower than that in the other solvents, which means viscosity play a minor role in the dimer yield, indicating the large angle rotation of the excited state between the two parts should be excluded.14a The existence of 1,1′,3,3′-tetramethyl group can inhibit large rotation and further confirms this speculation. The quenching fluorescence of the dimer is probably due to the β–β bond stretching vibration between the ground state and the excited state.18 Taking into account that the emission efficiency of the dimers is very sensitive to the solvent polarity, these properties of the dimers could be utilized for probing solvent polarity.19
3.3. Solid-state photophysical properties
Drop casted films were prepared by evaporation of the PMMA/dyes onto a quartz plate, which was utilized to measure the electronic spectra . The absorption bands of 5–6 are shifted to the red region in the solid compared to in solution with a broader shape, the broader and structureless bands can be attributed to strong intermolecular interactions in the solid-state (Fig. 2). Absorption spectra of 5 and 6 in the pure film are red-shifted by 21 nm and 9 nm relative to in CH2Cl2, respectively, indicating the introduction of the triphenylsilylphenyl group is effective to inhibit intermolecular aggregation.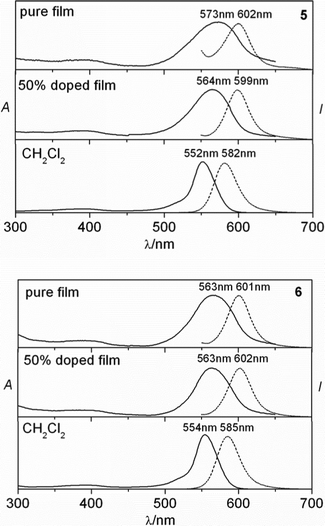 | ||
| Fig. 2 Absorption and fluorescent spectra of 5 and 6 in dichloromethane, doped film and pure film. | ||
The absolute quantum yields (ΦF) of the dyes in powder were measured by Edinburgh photonics absolute PL quantum yield measurement system FLS920 (Table 2). BODIPY dimer 6 shows efficient solid-state fluorescence with a quantum yield of 9.7%, which is stronger than that of dimer 5, suggesting that the introduction of the bulky triphenylsilyl group is effective in enhancing the solid-state emission efficiency of BODIPY dyes. However, more intensive solid-state emission of the BODIPY monomers is observed compared to relevant BODIPY dimer, most likely due to the inner structure increasing vibrational relaxation channels leading to fluorescent quenching in the solid-state. The result is consistent with the fluorescent behavior in solvent. Previously, Dreuw and our group showed that molecular properties of the individual molecules are preserved in the solid state.6,20 This result confirms our proposed suggestion, the inner structure factors and strong fluorescence in solvent with great oscillator strength (S0-S1) should be considered in designing solid-state emission BODIPY derivatives.
| Dyes | 3 | 4 | 5 | 6 |
|---|---|---|---|---|
| Φ F | 7.1% | 25.3% | 3.4% | 9.7% |
3.4. Molecular orbits and transition
To investigate the influence of structural modification with the bulky group and dimer on the spectroscopic and electronic properties, geometry optimizations were performed using density functional theory (DFT) employing a B3LYP functional and a 6-31G(d) basis. TD-DFT calculations were carried out with the same basis set. Similar to the calculated spectra of 4 and other reported BODIPY system,21 the calculated absorption spectra in the gas phase shows a blue shift and their sequence agrees well with the measured absorption spectra in solution. For dimers 5–6, HOMO and HOMO−1, LUMO and LOMO+1 show identical electron distribution with a small energy gap, the frontier orbital of the dimers can be considered the reorganization frontier orbital of two monomers with weak orbital interactions (Fig. 3). The main theoretical bands are composed of S0–S1 and S0–S3 transitions from the HOMO and HOMO−1/−2 to the LUMO and the LUMO+1, which can be assigned to the experimental absorption maximum band (Table 3). The frontier molecular orbital diagram exhibits destabilization of the HOMO and stabilization of the LUMO compared to those of the monomers, which is in good agreement with the results of the UV-vis measurements.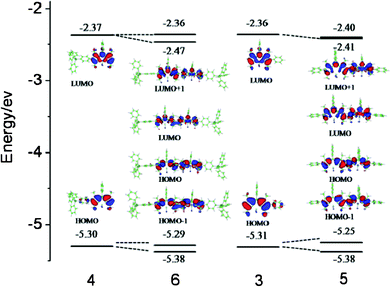 | ||
| Fig. 3 The frontier orbital energy of the dyes 3–6 were calculated with B3LYP level 6-31G(d) basis sets. | ||
| Statea | Energy [eV] | λ [nm] | ƒb | Orbitals (coefficient)c | |
|---|---|---|---|---|---|
| a Excited state. b Oscillator strength. c MOs involved in the transitions. | |||||
| 3 | S1 | 2.87 | 432 | 0.492 | H–L (0.550) H-1–L (−0.282) |
| S2 | 3.29 | 377 | 0.227 | H–L (0.218) H-1–L (0.605) | |
| 4 | S1 | 2.85 | 434 | 0.610 | H–L (0.552) H-1–L (−0.280) |
| S2 | 3.28 | 377 | 0.257 | H–L (0.215) H-1–L (0.604) | |
| 5 | S1 | 2.48 | 501 | 0.191 | H–L+1 (0.602) H-1–L (−0.332) |
| S2 | 2.50 | 497 | 0.001 | H–L (0.565) H-1–L+1 (−0.418) | |
| S3 | 2.71 | 458 | 1.364 | H–L+1 (0.264) H-1–L (0.561) H-2–L (−0.102) H-3–L+1 (−0.134) | |
| S5 | 3.24 | 383 | 0.327 | H-2–L (0.479) H-3–L+1 (0.433) | |
| 6 | S1 | 2.48 | 499 | 0.419 | H–L (0.623) H-1–L+1 (0.275) |
| S2 | 2.52 | 491 | 0.001 | H–L+1 (0.494) H-1–L (0.509) | |
| S3 | 2.74 | 453 | 1.475 | H–L (-0.180) H-1–L+1 (0.585) H-2–L (−0.120) | |
| S5 | 3.22 | 384 | 0.405 | H-2–L (0.458) H-3–L (−0.307) H-3–L+1 (0.247) H-2–L+1 (0.210) H-1–L+1 (0.111) | |
4. Conclusion
The synthesis, characterization, and theoretical analysis of BODIPY dimers have been investigated. Comparison with the optical properties of the corresponding monomer in solvent shows the β–β linked dimers exhibit higher molar absorption coefficients, lower fluorescent quantum yields and lifetime, increasing non-radiative rate constants and redshifted wavelengths. The luminescence yields of the dimers are dependent on solvent polarity and decrease dramatically in acetonitrile. The introduction of a triphenylsilylphenyl group as a bulky group is clearly interesting in the foundation of new BODIPY systems with efficient solid-state emission. Triphenylsilylphenyl substituted BODIPY monomer and dimer exhibit effective solid-state emission which are more intensive than those of the phenyl substituted monomers and dimers, respectively, most likely due to the introduction of the bulky group to inhibit aggregation. Meanwhile, the weak solid-state emission of the BODIPY dimers is observed compared to relevant BODIPY monomers, probably due to inner structural factors increasing vibrational relaxation channels. The absorbance maxima of the dimers is red-shifted around 40 nm with respect to the relevant monomer, indicating the oxidative coupling approach is an effective way for shifting the wavelength to the red region.Acknowledgements
We are thankful to the NSFC (nos. 21101049 and 21101050), the Zhejiang Provincial Natural Science Foundation of China (Y4110297) and the innovation teams for organosilicon chemistry (2009R50016) for their financial support.References
- A. Treibs and F. H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–213 CrossRef CAS.
- (a) A. B. Descalzo, H. J. Xu, Z. Shen and K. Rurack, Ann. N. Y. Acad. Sci., 2008, 1130, 164–171 CrossRef CAS; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS; (c) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS; (d) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- (a) Y. Ooyama, T. Okamoto, T. Yamaguchi, T. Suzuki, A. Hayashi and K. Yoshida, Chem.–Eur. J., 2006, 12, 7827–7838 CrossRef CAS; (b) M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS.
- (a) A. Hepp, G. Ulrich, R. Schmechel, H. von Seggern and R. Ziessel, Synth. Met., 2004, 146, 11–15 CrossRef CAS; (b) H. Langhals, O. Krotz, K. Polborn and P. Mayer, Angew. Chem., Int. Ed., 2005, 44, 2427–2428 CrossRef CAS.
- (a) T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildirim, A. Tulek, M. Bayindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105–2107 CrossRef CAS; (b) S. Badré, V. Monnier, R. Méallet-Renault, C. Dumas-Verdes, E. Y. Schmidt, A. I. Mikhaleva, G. Laurent, G. Georges Levi, A. Ibanez, B. A. Trofimov and R. B. J. Pansu, J. Photochem. Photobiol., A, 2006, 183, 238–246 CrossRef; (c) T. T. Vu, S. Badré, C. Dumas-Verdes, J. J. Vachon, C. Julien, P. Audebert, E. Y. Senotrusova, E. Y. Schmidt, B. A. Trofimov, R. B. Pansu, G. Clavier and R. Méallet-Renault, J. Phys. Chem. C, 2009, 113, 11844–11855 CrossRef CAS; (d) Y. Kubota, J. Uehara, K. Funabiki, M. Ebihara and M. Matsui, Tetrahedron Lett., 2010, 51, 6195–6198 CrossRef CAS; (e) D. Zhang, Y. Wen, Y. Xiao, G. Yu, Y. Liu and X. Qian, Chem. Commun., 2008, 4777–4779 RSC.
- H. Lu, Q. Wang, L. Gai, Z. Li, Y. Deng, X. Xiao, G. Lai and Z. Shen, Chem.–Eur. J., 2012, 18, 7852–7861 CrossRef CAS.
- S. Rihn, M. Erdem, A. De Nicola, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 1916–1919 CrossRef CAS.
- (a) A. B. Nepomnyashchii, M. Bröring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 8633–8645 CrossRef CAS; (b) A. B. Nepomnyashchii, M. Bröring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 19498–19504 CrossRef CAS.
- J. Olmsted, J. Phys. Chem., 1979, 83, 2581–2584 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Gaussian 03 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, J. T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211–10220 CrossRef CAS; (b) W. Qin, M. Baruah, M. V. Auweraer, F. C. De Schryver and N. Boens, J. Phys. Chem. A, 2005, 109, 7371–7384 CrossRef CAS.
- (a) B. Ventura, G. Marconi, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428–438 RSC; (b) M. Bröing, R. Krüger, S. Link, C. Kleeberg, S. Köler, X. Xie, B. Ventura and L. Flamigni, Chem.–Eur. J., 2008, 14, 2976–2983 CrossRef.
- (a) H. Lu, S. Shimizu, J. Mack, Z. Shen and N. Kobayashi, Chem.–Asian J., 2011, 6, 1026–1037 CrossRef CAS; (b) Z. Shen, H. Röhr, K. Rurack, H. Uno, M. Spieles, B. Schulz, G. Reck and N. Ono, Chem.–Eur. J., 2004, 10, 4853–4871 CrossRef CAS; (c) A. B. Descalzo, H. J. Xu, Z. L. Xue, K. Hoffmann, Z. Shen, M. G. Weller, X. Z. You and K. Rurack, Org. Lett., 2008, 10, 1581–1584 CrossRef CAS; (d) Y. Chen, J. Zhao, L. Xie, H. Guo and Q. Li, RSC Adv., 2012, 2, 3942–3953 RSC.
- (a) J. Bañuelos-Prieto, A. R. Agarrabeitia, I. García-Moreno, I. López-Arbeloa, A. Costela, L. Infantes, M. E. Perez-Ojeda, M. Palacios-Cuesta and M. J. Ortiz, Chem.–Eur. J., 2010, 16, 14094–14105 CrossRef; (b) M. J. Ortiz, I. Garcia-Moreno, A. R. Agarrabeitia, G. Duran-Sampedro, A. Costela, R. Sastre, F. López Arbeloa, J. Bañuelos Prieto and I. López Arbeloa, Phys. Chem. Chem. Phys., 2010, 12, 7804–7811 RSC.
- (a) Y. Hayashi, S. Yamaguchi, W. Y. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992–2995 CrossRef CAS; (b) F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001–10017 CrossRef CAS.
- X. Gao, Q. Peng, Y. Niu, D. Wang and Z. Shuai, Phys. Chem. Chem. Phys., 2012 10.1039/c2cp40347a.
- J. Bañuelos, I. J. Arroyo-Córdoba, I. Valois-Escamilla, A. Alvarez-Hernández, E. Peña-Cabrera, R. Hu, B. Z. Tang, I. Esnal, V. Martnez and I. López Arbeloa, RSC Adv., 2011, 1, 677–684 RSC.
- (a) A. Dreuw, J. Plötner, L. Lorenz, J. Wachtveitl, J. E. Djanhan, J. Brüning, T. Metz, M. Bolte and M. U. Schmidt, Angew. Chem., Int. Ed., 2005, 44, 7783–7786 CrossRef CAS; (b) J. Plötner and A. Dreuw, Phys. Chem. Chem. Phys., 2006, 8, 1197–1204 RSC.
- (a) B. L. Guennic, O. Maury and D. Jacquemin, Phys. Chem. Chem. Phys., 2012, 14, 157–164 RSC; (b) Y. W. Wang, A. B. Descalzo, Z. Shen, X. Z. You and K. Rurack, Chem.–Eur. J., 2010, 16, 2887–2903 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, calculated absorption, Ms and NMR spectra. See DOI: 10.1039/c2ra21040a |
| This journal is © The Royal Society of Chemistry 2012 |