Synthetic studies on pactamycin, a potent antitumor antibiotic†
Nobuyuki
Matsumoto
,
Takashi
Tsujimoto
,
Atsuo
Nakazaki
,
Minoru
Isobe
and
Toshio
Nishikawa
*
Graduate School of Bioagricultural Sciences, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: nisikawa@agr.nagoya-u.ac.jp
First published on 27th July 2012
Abstract
Two synthetic approaches to the functionalized cyclopentane core structure of pactamycin are described. The first approach employed the Pauson–Khand reaction of an enyne prepared from diacetone-D-glucose as a chiral starting material, providing a potentially useful tricyclic intermediate for pactamycin. The second approach involved an intramolecular 1,3-dipolar cycloaddition between the nitrone and acetylene functionalities of a precursor derived from an intermediate of the first approach. The expected isoxazoline cycloadduct was not detected, but instead an aziridine aldehyde was obtained, probably through ring contraction (Baldwin rearrangement) of the expected isoxazoline product. Further transformation provided a highly functionalized cyclopentane bearing an aniline at the C-3 position, but with different stereochemistry from that of pactamycin.
Introduction
Pactamycin (1) was isolated as a potent antitumor and antibacterial antibiotic from a fermentation broth of Streptomyces pactum var. pactum by researchers at Upjohn in 1962.1 The structure was initially elucidated by chemical degradation and spectroscopic methods in 1970,2 however, the originally proposed structure was revised to that shown in Fig. 1 by X-ray crystallographic analysis in 1972.3 The structure includes a highly functionalized cyclopentane, in which all the ring carbons are asymmetric centers bearing oxygen or nitrogen functionalities such as hydroxyl groups, an amine, m-acetylaniline, and dimethylurea. To our knowledge, pactamycin (1) and its analogue pactamycate (2) are the most structurally complex cyclopentane-containing natural products. Despite potent antitumor activities in vitro and in vivo,4 high toxicity has prohibited the development of a chemotherapeutic drug from this compound. Since the mechanism of action for pactamycin is the inhibition of protein synthesis5 through inhibition of the 30S ribosomal subunit,6 this compound has been employed as an inhibitor of protein synthesis in the field of biochemistry. The details of the bound structure with the 30S ribosomal subunit were recently disclosed by X-ray crystallography of the co-crystal between pactamycin and the protein.7 The challenging structure and the potent biological profile of pactamycin elicited our interest in its synthesis as well as its naturally occurring analogues, such as pactamycate,8 7-deoxypactamycin,9 and jogyamycin,10 in addition to unnatural derivatives. Surprisingly, little synthetic work on pactamycin had been reported11 until a report from this laboratory in 2005.12 Knapp reported their synthetic studies on pactamycin in 2007. In 2011, the first total synthesis of pactamycin was reported by Hanessian and co-workers.13 Quite recently two different approaches were disclosed by Johnson and Looper, independently.14,15 We describe herein the details of our efforts towards the total synthesis of pactamycin: (i) synthesis of a tricyclic intermediate possessing all the necessary carbons for the cyclopentane core of pactamycin by an intramolecular Pauson–Khand reaction as a key reaction,16 and (ii) an alternative approach based on an intramolecular 1,3-dipolar cycloaddition, which led to an advanced intermediate for pactamycin.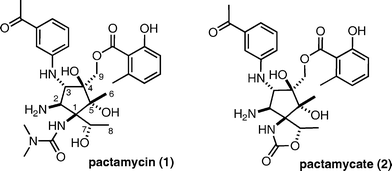 | ||
| Fig. 1 Structures of pactamycin (1) and pactamycate (2). | ||
The initial synthetic plan for pactamycin and its analogues17
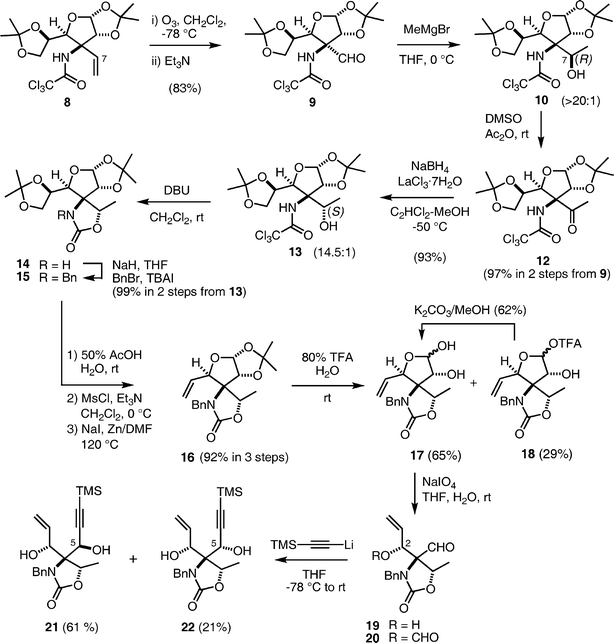
Our initial synthetic plan using an intramolecular Pauson–Khand reaction18 as a key reaction is illustrated in Scheme 1. Synthesis of the core structure of pactamycin was envisioned from tricyclic compound A through the following functional group transformations: (i) the amino group at the C-2 position would be installed by nucleophilic substitution of an ammonia equivalent such as azide in an SN2 manner; (ii) the nitrogen function at the C-3 position would be introduced by the Curtius rearrangement of a carboxylic acid, which could be obtained from oxidative cleavage of the silyl enol ether; (iii) the methyl group at the C-5 position would be installed by addition of a methyl anion to the ketone; and (iv) dimethylurea would be prepared by addition of dimethylamine to the cyclic carbamate. Since the enol ether A would be obtainable from the corresponding keto form B, compound B was regarded as an important intermediate. The cyclopentenone B would be accessible through an intramolecular Pauson–Khand reaction16 of enyne C, although the stereochemistry at the C-3 position of the product B could not be predicted. Since the acetylenic moiety could be introduced by addition of acetylide to an aldehyde and the vinyl group would be prepared from a diol, the enyne C was retrosynthesized into bis-acetonide D. The nitrogen function connected to the quaternary carbon center at the C-1 position would be introduced as a trichloroacetamide by the Overman rearrangement19 of the known exo-allylic alcohol E.20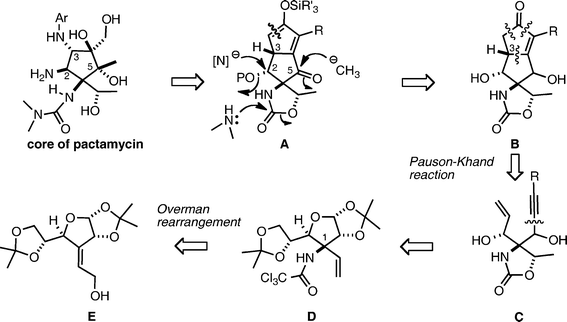 | ||
| Scheme 1 Initial synthetic plan for pactamycin. | ||
Synthesis of important intermediate B
Synthesis of the tricyclic compound B commenced with preparation of allylic alcohol E (6) from commercially available diacetone-D-glucose (3) according to Tadano's procedure20 (Scheme 2). This sequence is quite reproducible, however, oxidation of 3 on a 25–50 g scale required a significant amount of toxic PCC and a tedious work-up. Thus, we instead employed the Ley oxidation21 (TPAP (0.1 mol%) and NMO (2 equiv.) in the presence of 4 Å MS) for this purpose, although the yield was lower than for the PCC oxidation. Since the ketone was obtained as a mixture with its hydrate, the crude product was used for the subsequent Wittig reaction without any purification. Reaction of the resulting ketone with the Wittig reagent 4 in refluxing toluene afforded a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mixture of E/Z isomeric products 5a and 5b, from which the desired Z-product 5b was easily isolated by crystallization and column chromatography. The overall yield of 5b from 3 was about 50%. Reduction of the ester 5b with DIBAL-H gave the allylic alcohol 6, which was employed for the Overman rearrangement. The first issue of this synthetic route was the feasibility of the Overman rearrangement from 6, because two previous reports indicated that the [3,3]-sigmatropic rearrangement of the imidate 7 did not proceed.22 To our delight, improved conditions (addition of K2CO3 as an acid scavenger)23 for facile Overman rearrangement developed in our laboratory facilitated the sigmatropic rearrangement of 7. The allylic trichloroacetimidate 7 was prepared by treatment of 6 with Cl3C–CN and DBU in CH2Cl2, and then was exposed to the improved conditions for the Overman rearrangement: a xylene solution of 7 was refluxed in the presence of K2CO3 to afford 8 as a single product, in 84% overall yield in three steps from 5b. This success demonstrates the general usefulness of the improved conditions for the Overman rearrangement. The configuration of the newly formed chiral center was confirmed by observation of a NOESY correlation between protons Ha and Hb as shown in compound 8. The stereochemical outcome was consistent with the result of the Claisen rearrangement of an orthoester derived from 6, and can thus be rationalized by the transition state conformation depicted in 7. On the other hand, the Overman rearrangement of the imidate derived from the minor product 5a under identical conditions gave a complex mixture of products, including a small amount of the desired product 8. Therefore, separation of the regioisomers 5a and 5b after the Wittig reaction was best for easy purification of 8 on a multi-gram scale. In practice, we synthesized about 20 g of 8 from 30 g of 3 by the sequence illustrated in Scheme 2. The multi-functionality of 8 and its easy preparation highlights its versatility as a useful intermediate for the synthesis of nitrogen-containing natural and unnatural compounds.
:5 mixture of E/Z isomeric products 5a and 5b, from which the desired Z-product 5b was easily isolated by crystallization and column chromatography. The overall yield of 5b from 3 was about 50%. Reduction of the ester 5b with DIBAL-H gave the allylic alcohol 6, which was employed for the Overman rearrangement. The first issue of this synthetic route was the feasibility of the Overman rearrangement from 6, because two previous reports indicated that the [3,3]-sigmatropic rearrangement of the imidate 7 did not proceed.22 To our delight, improved conditions (addition of K2CO3 as an acid scavenger)23 for facile Overman rearrangement developed in our laboratory facilitated the sigmatropic rearrangement of 7. The allylic trichloroacetimidate 7 was prepared by treatment of 6 with Cl3C–CN and DBU in CH2Cl2, and then was exposed to the improved conditions for the Overman rearrangement: a xylene solution of 7 was refluxed in the presence of K2CO3 to afford 8 as a single product, in 84% overall yield in three steps from 5b. This success demonstrates the general usefulness of the improved conditions for the Overman rearrangement. The configuration of the newly formed chiral center was confirmed by observation of a NOESY correlation between protons Ha and Hb as shown in compound 8. The stereochemical outcome was consistent with the result of the Claisen rearrangement of an orthoester derived from 6, and can thus be rationalized by the transition state conformation depicted in 7. On the other hand, the Overman rearrangement of the imidate derived from the minor product 5a under identical conditions gave a complex mixture of products, including a small amount of the desired product 8. Therefore, separation of the regioisomers 5a and 5b after the Wittig reaction was best for easy purification of 8 on a multi-gram scale. In practice, we synthesized about 20 g of 8 from 30 g of 3 by the sequence illustrated in Scheme 2. The multi-functionality of 8 and its easy preparation highlights its versatility as a useful intermediate for the synthesis of nitrogen-containing natural and unnatural compounds.
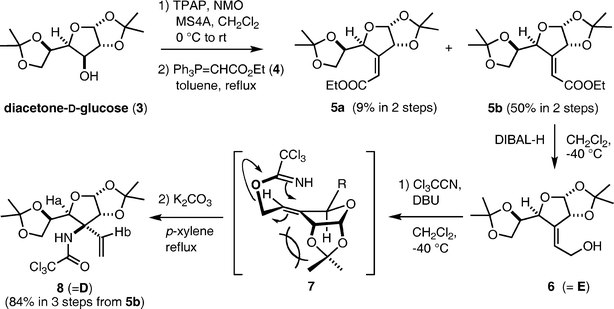 | ||
| Scheme 2 Preparation of trichloroacetamide 8. | ||
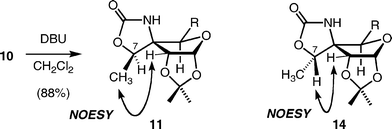
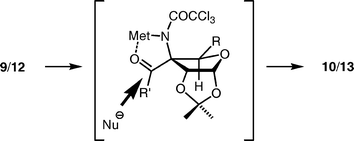
Synthesis of the precursor C for the Pauson–Khand reaction from the trichloroacetamide 8 (D) is shown in Scheme 3. Our initial attempts to hydroxylate the C-7 position via a neighbouring group participation of the trichloroacetamide24 failed. We therefore synthesized this moiety by ozonolysis of the vinyl group followed by addition of the methyl group.25 Ozonolysis of 8 was carried out in CH2Cl2 at −78 °C and quenched with Et3N26 to afford aldehyde 9 in good yield. Addition of MeMgBr to 9 in THF gave alcohol 10 with the undesired (R)-configuration in a highly stereoselective manner (>20:1 by 1H-NMR).27 The R-configuration at the C-7 position was determined by analysis of the NOESY spectra of cyclic carbamate 11 obtained by treatment of 10 with DBU (Fig. 2). The high stereoselectivity could be explained by the preferential attack of the methyl anion from the less hindered face of the magnesium ion-chelated intermediate (Fig. 3, Met = Mg2+, R′ = H, Nu = Me−). This consideration led us to examine inversion of configuration at the C-7 position by oxidation of the alcohol followed by hydride reduction, because a similar chelation mechanism could operate in the reduction as shown in Fig. 3 (R′ = Me, Nu = H−). Thus, 10 was oxidized with DMSO–Ac2O (Albright–Goldman oxidation),28 and then reduction of the resulting ketone 12 was investigated. Treatment of 12 with NaBH4 in MeOH caused significant deprotection of the trichloroacetyl group,29 however, the major product obtained was (S)-isomer 13, as expected. After extensive examination, we found that Luche reduction,30 using NaBH4 and LnCl3·7H2O in MeOH at −50 °C, best afforded the desired (S)-product 14 in the highest stereoselectivity (13:10 = 14.5:1 by 1H-NMR).31 The (S)-configuration was confirmed by NOESY spectra of the cyclic carbamate 14 (Fig. 2) derived from 13 by treatment with DBU in CH2Cl2.
 | ||
| Fig. 2 Stereochemical assignment of compounds 11 and 14. | ||
 | ||
| Scheme 3 Synthesis of precursors 21 and 22 for the Pauson–Khand reaction. | ||
The cyclic carbamate 14 was benzylated, and the two acetonides of the product 15 were then elaborated to vinyl and aldehyde groups as follows. The terminal acetonide of 15 was selectively hydrolyzed in 50% aqueous acetic acid to the diol, which was converted to the vinyl group of 16 in two steps, which included methanesulfonylation followed by reduction with NaI and Zn in DMF at 120 °C (Tipson–Cohen procedure).32 The remaining acetonide of the product 16 was deprotected with 80% aqueous TFA to give a mixture of diol 17 and its trifluoroacetate 18, which was hydrolyzed with K2CO3 in MeOH; thus, an 83% combined yield of 17 was obtained from 16. In an early stage of this study,1217 was reduced with NaBH4 followed by oxidative cleavage with sodium periodate to afford aldehyde 19, which was used for the next reaction. However, in a multi-gram scale synthesis of 19, reduction of 17 with NaBH4 proved to be problematic, because a stable cyclic borate was formed under the reaction conditions, and the hydrolysis was difficult. We therefore examined an alternative route to aldehyde 19 for the addition of the acetylide: 17 was treated with NaIO4 to give the aldehyde 20 bearing a formate group at the C-2 position.33 When formate 20 was employed as a substrate for the addition of lithium acetylide, prepared from TMS acetylene with n-BuLi, a mixture of adducts was obtained in 90% yield with a moderate diastereoselectivity (21: 61%, 22: 21%). The configuration at the C-5 position of the major product 21 was determined by NMR experiments on the products from the subsequent Pauson–Khand reaction (vide infra). This stereoselectivity may be attributed to a preferential attack of the acetylide from the less hindered face of a chelated intermediate, as shown in Fig. 4.
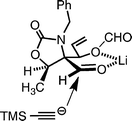 | ||
| Fig. 4 Proposed mechanism for addition of lithium acetylide to aldehyde 20. | ||
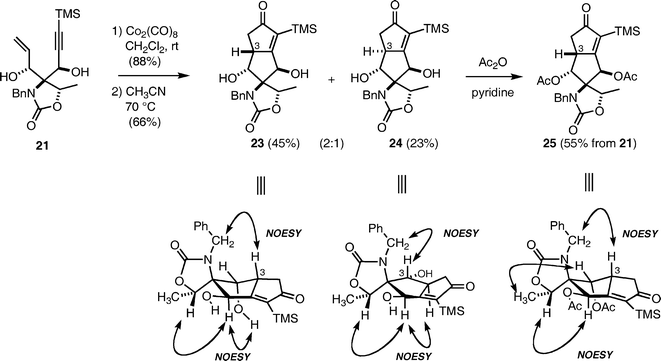
With enyne 21 in hand, the Pauson–Khand reaction was next investigated (Scheme 4). Enyne 21 was treated with Co2(CO)8 to give the acetylene–cobalt complex in 88% yield, which underwent the intramolecular Pauson–Khand reaction in acetonitrile at 70 °C to afford a diastereomeric mixture of 23 and 24 in a nearly 2:1 ratio (by 1H NMR). Interestingly, the corresponding diacetate and bisTMS ether of 21 and the diastereomer 22 did not undergo the Pauson–Khand reaction under identical conditions. In our preliminary report,12 we described the products 23 and 24 as inseparable, but acetylation of the mixture enabled the isolation of 25 for structure determination by NMR techniques. However, through further careful experiments, we found that compounds 23 and 24 were separable by flash chromatography eluting with hexane–acetone (4:1). Extensive analysis of the NMR spectra, including NOESY spectra of 23, 24, and 25, elucidated the configurations at the C-3 position as shown in Scheme 4. The tricyclic compound 23 was recognized as the important intermediate B in our initial plan (Scheme 1), however, transformation of 23 to enol ether A proved to be difficult, because of its base sensitivity. We therefore revised the initial synthetic plan as shown in Scheme 5.
 | ||
| Scheme 4 Intramolecular Pauson–Khand reaction of enyne 21. | ||
 | ||
| Scheme 5 Revised synthetic plan. | ||
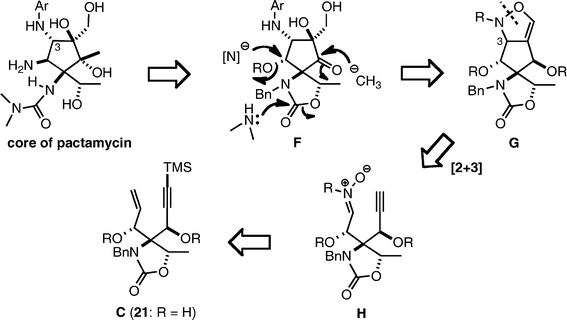
The revised synthetic plan
We planned to utilize an intramolecular 1,3-dipolar cycloaddition between the nitrone and acetylene34 of H as the key reaction for construction of the cyclopentane structure. The expected product G would be an isoxazoline-fused cyclopentane, which would possess suitable functional groups for the synthesis of pactamycin. Although the stereochemistry of the product could not be predicted, installation of a nitrogen functionality at the C-3 position simultaneously with the construction of the cyclopentane ring was attractive (Scheme 5). The precursor H could be prepared from enyne C (21), the same precursor for the Pauson–Khand reaction as described above.Synthesis toward cyclopentane F through the 1,3-dipolar cycloaddition
Desilylation of 21 with K2CO3 in MeOH was followed by benzylation of the two hydroxyl groups under conventional conditions to give 26 in 78% overall yield in two steps (Scheme 6). The vinyl group of the product 26 was cleaved by ozonolysis, yielding aldehyde 27, which was condensed with N-hydroxyaniline to afford nitrone 28 as a precursor for the intramolecular 1,3-dipolar cycloaddition. The [2 + 3] cycloaddition was carried out at reflux in toluene to afford a single product. However, analysis of the NMR spectra revealed that the structure of the product was not the expected isoxazoline 29, but rather the aziridine aldehyde 30. This product might be formed through ring contraction (Baldwin rearrangement)35,36 of the expected isoxazoline 29, although 29 was not detected at all.37,38 Formation of the aziridine aldehyde 30 was unexpected and its stereochemistry could not be determined at this stage. However, we employed 30 in further synthetic transformations, because its functionality seemed to be more suitable than that of 29 for the approach to pactamycin.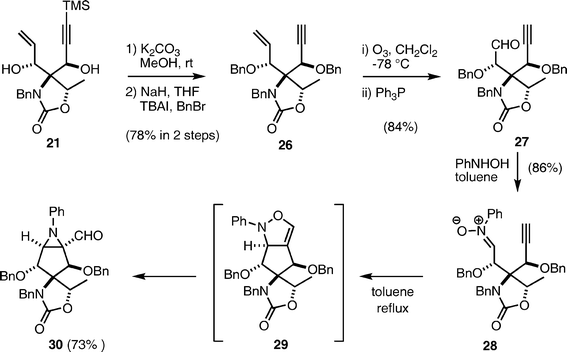 | ||
| Scheme 6 Synthesis of aziridine aldehyde 30. | ||
Thus, the aldehyde of 30 was reduced with NaBH4 and the resulting alcohol was transformed into the corresponding iodide 32 in two steps (methanesulfonylation with MsCl and Et3N followed by treatment with TBAI (n-Bu4NI) in THF at 60 °C). Upon exposure of the aziridine 32 to TBAI in the presence of BF3·OEt2 in CH2Cl2 at 0 °C, ring opening of the aziridine proceeded to provide allylamine 33 in 79% yield. Addition of a stoichiometric amount of BF3·OEt2 was indispensable for obtaining a reproducible result. Dihydroxylation of the product 33 with a catalytic amount of OsO4 and NMO proceeded in a highly stereoselective manner to furnish diol 34 as a single diastereomer in moderate yield. The stereochemistry of 34 was determined by extensive NMR analysis, as shown in Scheme 7, which in turn clarified the stereochemistry of aziridine 30. Unfortunately, the configurations of C-3 and C-4 were opposite to those of pactamycin.
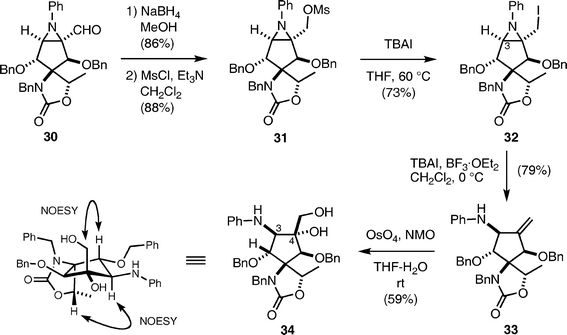 | ||
| Scheme 7 | ||
The high stereoselectivity observed in the intramolecular 1,3-dipolar cycloaddition of 2839 might be rationalized by comparing the stabilities of two possible transition state conformations, TS-1 and TS-2 (Scheme 8). The TS-1 conformation is seemingly more stable than TS-2, because TS-2 includes two axial-like benzyloxy substituents at the C-5 and C-2 positions. In addition, repulsion between the methyl at C-8 and the benzyloxy group at C-2 in TS-2 was more pronounced than in TS-1. These steric effects might result in the exclusive formation of aziridine aldehyde 30via isoxazoline 29.
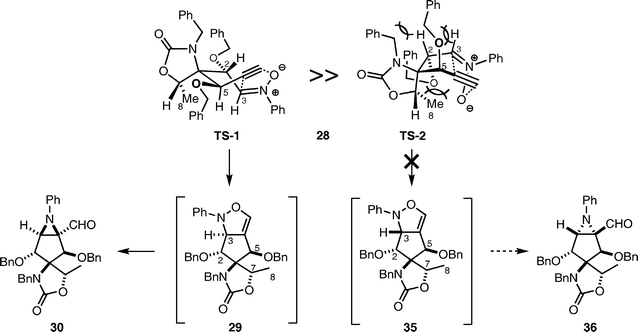 | ||
| Scheme 8 Proposed mechanism for stereoselective 1,3-dipolar cycloaddition of 28. | ||
In summary, we have described the synthesis of highly functionalized cyclopentane analogues of pactamycin by two different approaches. The first approach by the Pauson–Khand reaction provided a tricyclic cyclopentane fused with a cyclopentenone; however, further transformation proved to be difficult because of base sensitivity. The second approach consisted of an intramolecular 1,3-dipolar cycloaddition followed by a spontaneous Baldwin rearrangement to give an isoxazoline-fused cyclopentane, which was elaborated to a highly functionalized cyclopentane with different stereochemistry to that of naturally occurring pactamycin. These compounds should be useful in the study of pactamycin structure–activity relationships. On the basis of these results, further synthetic studies toward the total synthesis of pactamycin are now in progress in our laboratory.
Experimental
General
Melting points (mp) were recorded on a Yanaco MP-S3 melting point apparatus and are not corrected. Optical rotations were measured on a JASCO DIP-370 digital polarimeter. Infrared spectra (IR) were recorded on a JASCO FT/IR-8300 spectrophotometer and are reported in wave number (cm−1). Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AVANCE-400 (400 MHz) or a Varian Gemini-2000 (300 MHz) spectrometer. NMR samples were dissolved in CDCl3, CD3OD or C6D6, and chemical shifts are reported in ppm relative to TMS or the residual undeuterated solvent (CD3OD as δ = 3.31, C6D6 as δ = 7.16). 1H NMR data are reported as follows; chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broadened, m = multiplet), coupling constant, and assignment. Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker AVANCE-400 (100 MHz) or a Varian Gemini-2000 (75 MHz) spectrometer. The samples were dissolved in CDCl3, and chemical shifts are reported in ppm relative to the solvent (CDCl3 as δ = 77.0). 1H NMR and 13C NMR spectra were measured at 300 K unless otherwise noted. Elemental analyses were performed at the Analytical Laboratory of the Graduate School of Bioagricultural Sciences, Nagoya University. High resolution mass spectra (HRMS) were recorded on an Applied Biosystems Mariner Biospectrometry Workstation and are reported in m/z. Reactions were monitored by thin-layer chromatography (TLC) on 0.25 mm silica gel coated glass plate 60 F254 (Merck, #1.05715). Silica gel 60 (particle size 63–200 μm, 70–230 mesh ASTM, Merck Ltd.) and silica gel 60 N (spherical, neutral, particle size 63–210 μm, Kanto Chemical Co., Inc.) were used for open-column chromatography. Flash silica gel 60 N (spherical, neutral, particle size 40–50 μm, Kanto Chemical Co., Inc.) was used for flash-column chromatography. Preparative thin-layer chromatographic separations were carried out on 0.5 mm silica gel coated glass plate 60 F254 (Merck, #1.05744). Unless otherwise noted, non-aqueous reactions were carried out in oven-dried (120 °C) or flame-dried glassware under nitrogen or argon. Dry CH3CN was distilled from CaH2. Dry THF and CH2Cl2 were purchased from Kanto Chemical Co., Inc. All other commercially available reagents were used as received.Oxidation of diacetone-D-glucose (3)
To a solution of diacetone-D-glucose 3 (30.0 g, 115 mmol) in dry CH2Cl2 (600 mL) was added 4 Å MS (57.6 g) and stirring was continued for 30 min at 0 °C under nitrogen. NMO (27.0 g, 230 mmol) and TPAP (4.04 g, 11.5 mmol) were subsequently added. The mixture was stirred for 10 min at 0 °C, and then for an additional 20 min at room temperature. The mixture was filtered through a pad of Super-Cel, and the precipitate was rinsed with Et2O. The combined filtrate was passed through a column packed with anhydrous Na2SO4 and silica gel (250 g) eluting with Et2O, and concentrated to dryness in vacuo. The residue (19.6 g) was used for the next reaction without purification.Unsaturated esters 5a and 5b
To a solution of the crude product (19.6 g) in toluene (760 mL) was added Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et (4, 27.8 g, 79.8 mmol). The mixture was heated to reflux temperature for 5 h with vigorous stirring. After cooling to room temperature, the mixture was concentrated to dryness in vacuo. The residue was passed through a column (silica gel 300 g) eluting with hexane–Et2O (1:1) for removal of Ph3PO, and concentrated to dryness in vacuo. The crude product was purified by column chromatography (silica gel 400 g) eluting with hexane–AcOEt (7:1) to give Z-ester 5b (18.8 g, 50% in 2 steps) as a white solid and E-ester 5a (3.46 g, 9% in 2 steps) as a colorless oil.
CHCO2Et (4, 27.8 g, 79.8 mmol). The mixture was heated to reflux temperature for 5 h with vigorous stirring. After cooling to room temperature, the mixture was concentrated to dryness in vacuo. The residue was passed through a column (silica gel 300 g) eluting with hexane–Et2O (1:1) for removal of Ph3PO, and concentrated to dryness in vacuo. The crude product was purified by column chromatography (silica gel 400 g) eluting with hexane–AcOEt (7:1) to give Z-ester 5b (18.8 g, 50% in 2 steps) as a white solid and E-ester 5a (3.46 g, 9% in 2 steps) as a colorless oil.
Allyl alcohol 6
To a solution of Z-ester 5b (18.8 g, 57.2 mmol) in dry CH2Cl2 (520 mL) at −40 °C was added DIBAL-H (0.94 M in hexane, 160 mL, 0.15 mol) dropwise under nitrogen. The mixture was stirred for 2 h at −40 °C, and then quenched by addition of 33% aqueous potassium sodium tartrate (300 mL). The combined mixture was stirred for 11 h at room temperature and then partitioned. The aqueous layer was extracted with CH2Cl2 (100 mL × 3). The combined organic layers were washed with H2O (100 mL) and brine (100 mL × 2), dried over Na2SO4, and concentrated to dryness in vacuo. The residue (16.3 g) was used for the next reaction without purification.
1H-NMR (300 MHz, CDCl3) δ 1.35 (3H, s, acetonide), 1.38 (3H, s, acetonide), 1.43 (3H, s, acetonide), 1.49 (3H, s, acetonide), 3.90–4.10 (3H, m), 4.28–4.42 (2H, m), 4.62–4.68 (1H, m), 5.24 (1H, d, J = 4.5 Hz, OCHCHC), 5.85 (1H, d, J = 4.5 Hz, OCHCHC), 6.12 (1H, tt, J = 6, 1.5 Hz, CCH).
Trichloroacetimidate 7
To a solution of the crude product 6 (16.3 g) in dry CH2Cl2 (550 mL) at −40 °C were added DBU (12.8 mL, 85.6 mmol) and CCl3CN (7.5 mL, 74.8 mmol) under nitrogen, and stirring was continued for 2 h at −40 °C. The mixture was quenched by addition of saturated aqueous NH4Cl (200 mL), and then stirred for 1 h at room temperature. After H2O (100 mL) was added, the mixture was extracted with CH2Cl2 (100 mL × 3). The combined organic layers were washed with saturated aqueous NH4Cl (100 mL), H2O (100 mL) and brine (100 mL × 2), dried over Na2SO4, and concentrated to dryness in vacuo. The residue (26.2 g) was used for the next reaction without purification.Trichloroacetamide 8
A mixture of the crude product 7 (23.2 g) and K2CO3 (1.2 g) in p-xylene was heated to reflux temperature for 84 h with vigorous stirring. After cooling to room temperature, the reaction mixture was filtered through a pad of Super-Cel, and the precipitate was rinsed with toluene (200 mL). The combined filtrate was concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 400 g) eluting with hexane–AcOEt (9:1) to give trichloroacetamide 8 (20.8 g, 84% in 3 steps) as a colorless oil.
[α]26D + 41.4 (c = 1.05, CHCl3). IR (film) νmax 3312, 2990, 1720, 1507, 1375, 1248, 1216, 1164, 1081, 1007, 872, 844, 822. 1H-NMR (300 MHz, CDCl3) δ 1.35 (3H, s, acetonide), 1.37 (3H, s, acetonide), 1.49 (3H, s, acetonide), 1.57 (3H, s, acetonide), 3.96 (1H, t, J = 8 Hz, CHaHbCHC), 4.06 (1H, dd, J = 8, 6.5 Hz, CHaHbCHCH), 4.18 (1H, d, J = 3.5 Hz, CHaHbCHCH), 4.56 (1H, ddd, J = 8, 6.5, 3.5 Hz, CHaHbCHCH), 5.28 (1H, d, J = 3.5 Hz, OCHCHC), 5.43 (1H, d, J = 11 Hz, CH=CHcHd) 5.44 (1H, d, J = 17.5 Hz, CH=CHcHd), 5.92 (1H, d, J = 3.5 Hz, OCHCHC), 6.04 (1H, dd, J = 17.5, 11 Hz, CH=CHcHd), 8.54 (1H, brs, NH). 13C-NMR (75 MHz, CDCl3) δ 24.8, 26.0, 26.5, 26.6, 66.0, 69.8, 75.4, 78.5, 83.7, 92.9, 104.0, 110.5, 112.5, 117.6, 131.0, 161.5. Elemental Analysis Calcdd for C16H22Cl3NO6: C, 44.62; H, 5.15; N, 3.25. found: C, 44.63; H, 5.12; N, 3.23%.
Aldehyde 9
A solution of trichloroacetamide 8 (12.8 g, 29.6 mmol) in CH2Cl2 (70 mL) was cooled to −78 °C under argon. Ozone was passed into the solution until the color of the solution became blue, and the reaction mixture was stirred for 12 h at the same temperature. After the ozone was purged with argon, Et3N (7.0 mL, 0.050 mol) was added at −78 °C, and the reaction mixture was stirred for 1 h at room temperature. The mixture was poured into 80% NH4Cl aqueous solution, and stirring was continued for 30 min. The aqueous layer was separated and extracted with CH2Cl2 (50 mL × 3). The combined organic extracts were washed with H2O (50 mL), brine (50 mL), and then dried over Na2SO4. The solution was concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 150 g) eluting with hexane–EtOAc (4:1) to give aldehyde 9 (10.7 g, 83%) as a white solid.
[α]25D + 34.0 (c = 1.00, CHCl3). mp: 113–116 °C (Hexane–AcOEt). IR (film) νmax 3357, 2989, 2886, 1724, 1510, 1375, 1214, 1073. 1H-NMR (300 MHz, CDCl3) δ 1.25 (3H, s, Me), 1.32 (3H, s, Me), 1.34 (3H, s, Me), 1.64 (3H, s, Me), 4.00–4.18 (3H, m), 4.60 (1H, d, J = 8.5 Hz, CHaHbCHCH), 5.19 (1H, d, J = 3.5 Hz, OCHCHC), 6.35 (1H, d, J = 3.5 Hz, OCHCHC), 7.88 (1H, br, NH), 9.69 (1H, s, CHO). 13C-NMR (75 MHz, CDCl3) δ 24.5, 25.8, 26.5, 26.7, 67.3, 72.1, 73.1, 81.6, 86.8, 92.4, 107.7, 109.9, 113.3, 161.5, 194.5. Elemental Analysis Calcdd for C15H20Cl3NO7: C, 41.67; H, 4.66; N, 3.24. found: C, 41.81; H, 4.79; N, 3.24%.
Alcohol 10
To a solution of aldehyde 9 (16.9 g, 39.1 mmol) in dry THF (400 mL) at 0 °C was added MeMgBr (0.99 M in THF, 100 mL, 99 mmol) under nitrogen. The reaction mixture was stirred for 2 h at 0 °C, and then quenched by addition of saturated aqueous NH4Cl (100 mL). After stirring for 30 min at room temperature, H2O (100 mL) was added. The mixture was extracted with Et2O (100 mL × 3). The combined organic layers were washed with H2O (100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The crude product (18.0 g) was used for the next reaction without purification. A part of the crude product was purified by preparative thin-layer chromatography for analytical purposes.[α]27D + 30.8 (c = 1.02, CHCl3). IR (film) νmax 3424, 3350, 2988, 1732, 1506, 1375, 1214, 1066, 820. 1H-NMR (400 MHz, CDCl3) δ 1.33 (3H, s, acetonide), 1.36 (3H, dd, J = 6, 0.5 Hz, CHCH3), 1.37 (3H, s, acetonide), 1.47 (3H, s, acetonide), 1.53 (3H, s, acetonide), 3.98 (1H, d, J = 9 Hz, CHHCHCH), 4.03–4.13 (2H, m), 4.17–4.26 (2H, m), 4.29 (1H, brs), 5.13 (1H, d, J = 3.5 Hz, OCHCHC), 6.22 (1H, d, J = 3.5 Hz, OCHCHC), 7.65 (1H, brs, NH). 13C-NMR (100 MHz, CDCl3) δ 17.5, 25.1, 26.0, 26.5, 26.6, 68.0, 68.3, 71.0, 71.8, 84.4, 85.4, 93.3, 105.9, 110.6, 111.9, 161.7. Elemental Analysis Calcdd for C16H24Cl3NO7: C, 42.83; H, 5.39; N, 3.12. found: C, 42.82; H, 5.31; N, 2.97%.
Carbamate 11
To a solution of alcohol 10 (21.3 mg, 47.5 μmol) in dry CH2Cl2 (1 mL) at 0 °C was added DBU (10 μL, 71.2 μmol) and stirring was continued for 4 h at room temperature under nitrogen. The mixture was quenched by addition of saturated aqueous NH4Cl (5 mL) and stirred for 30 min at room temperature. The mixture was partitioned, and the aqueous layer was extracted with CH2Cl2 (3 mL × 3). The combined organic layers were washed with H2O (3 mL) and brine (3 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by preparative thin-layer chromatography (hexane–AcOEt, 1:1) to give carbamate 11 (13.8 mg, 88%) as a colorless oil.
1H-NMR (400 MHz, CDCl3) δ 1.32 (3H, s, acetonide), 1.33 (3H, s, acetonide), 1.42 (3H, s, acetonide), 1.52 (3H, s, acetonide), 1.56 (3H, d, J = 6.5 Hz, CHCH3), 3.90–4.01 (2H, m), 4.09–4.17 (2H, m), 4.49 (1H, d, J = 3 Hz, OCHCHC), 4.89 (1H, q, J = 6.5 Hz, CHCH3), 5.78 (1H, d, J = 3 Hz, OCHCHC), 6.88 (1H, brs, NH). 13C-NMR (100 MHz, CDCl3) δ 17.6, 25.1, 26.4, 26.7, 27.0, 67.7, 70.6, 72.6, 75.8, 81.3, 83.7, 103.8, 109.9, 113.5, 159.0. HRMS (ESI) for C15H23NNaO7 (M+Na+), calcd 352.1372, found 352.1368.
Methyl ketone 12
To a solution of the crude product 10 (18.0 g) in DMSO (360 mL) at 0 °C was added Ac2O (45 mL). After stirring for 11 h at room temperature, the mixture was poured into an ice-cold mixture of saturated aqueous NaHCO3 (500 mL) and Et2O (100 mL), and stirring was continued for 1 h at ice bath temperature. The resulting precipitate was filtered off and washed with cold Et2O (200 mL) to give ketone (17.0 g, 97%, 2 steps) as a white solid. A part of the crude product was purified by preparative thin-layer chromatography for analytical purposes.[α]26D + 60.7 (c = 1.04, CHCl3). mp: 196–197 °C (Hexane/AcOEt). IR (film) νmax 3239, 2988, 1714, 1542, 1211, 1081, 1039, 855. 1H-NMR (400 MHz, CDCl3) δ 1.27 (3H, s, acetonide), 1.33 (3H, s, acetonide), 1.37 (3H, s, acetonide), 1.61 (3H, s, acetonide), 2.46 (3H, s, COCH3), 3.97 (1H, dd, J = 8.5, 5.5 Hz, CHHCHCH), 4.08 (1H, ddd, J = 8, 6, 5.5 Hz, CHHCHCH), 4.19 (1H, dd, J = 8.5, 6 Hz, CHHCHCH), 4.75 (1H, d, J = 8 Hz, CHHCHCH), 5.26 (1H, d, J = 3.5 Hz, OCHCHC), 6.26 (1H, d, J = 3.5 Hz, OCHCHC), 7.97 (1H, brs, NH). 13C-NMR (100 MHz, CDCl3) δ 24.7, 25.9, 26.0, 26.3, 28.9, 67.8, 73.5, 75.7, 83.2, 84.5, 92.9, 107.1, 109.9, 112.8, 161.3, 200.2. Elemental Analysis Calcdd for C16H22Cl3NO7: C, 43.02; H, 4.96; N, 3.14. found: C, 43.00; H, 4.81; N, 2.99%.
Alcohols 13 and 10
To a solution of the ketone 12 (7.24 g, 16.2 mmol) in MeOH–CH2Cl2 (1:1, 460 mL) was added LaCl3·7H2O (6.01 g, 16.2 mmol). The mixture was stirred for 30 min at room temperature, and then cooled to −50 °C. To this solution was added NaBH4 (1.23 g, 32.4 mmol) portionwise. After stirring for 5 h at −50 °C, saturated aqueous NH4Cl (100 mL) was added. Stirring was continued for additional 30 min at room temperature, H2O (100 mL) was added. The mixture was extracted with Et2O (100 mL × 3). The combined organic layers were washed with H2O (100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 250 g) eluting with hexane–AcOEt (6:1) to give the desired alcohol 13 (6.32 g, 87%) as a white solid and the undesired alcohol 10 (0.45 g, 6%) as a colorless oil.
[α]26D + 13.5 (c = 1.13, CHCl3). mp: 89–93 °C. IR (film) νmax 3461, 3329, 2987, 1731, 1523, 1375, 1213, 1167, 1070, 820. 1H-NMR (400 MHz, CDCl3) δ 1.35 (3H, s, acetonide), 1.36 (3H, s, acetonide), 1.48 (3H, s, acetonide), 1.52 (3H, d, J = 6.5 Hz, CHCH3), 1.58 (3H, s, acetonide), 3.27 (1H, d, J = 11.5 Hz, OH), 4.02 (1H, dd, J = 8.5, 6 Hz, CHHCHCH), 4.04–4.14 (2H, m, CHHCHCH and CHCH3), 4.23 (1H, dd, J = 8.5, 6 Hz, CHHCHCH), 4.42 (1H, d, J = 8.5 Hz, CHHCHCH), 5.43 (1H, d, J = 3.5 Hz, OCHCHC), 6.14 (1H, d, J = 3.5 Hz, OCHCHC), 7.52 (1H, brs, NH). 13C-NMR (100 MHz, CDCl3) δ 20.3, 25.0, 25.9, 26.2, 26.5, 68.4, 69.3, 71.3, 73.6, 80.3, 84.3, 93.2, 106.0, 110.2, 112.4, 161.3. Elemental Analysis Calcdd for C16H24Cl3NO7: C, 42.83; H, 5.39; N, 3.12. found: C, 42.81; H, 5.18; N, 3.15%.
Cyclic carbamate 14
To a solution of alcohol 13 (12.9 g, 28.7 mmol) in dry CH2Cl2 (300 mL) at 0 °C was added DBU (5.20 mL, 34.4 mmol) and stirring was continued for 2 h at room temperature under nitrogen. The mixture was quenched by addition of saturated aqueous NH4Cl (200 mL) and stirred for 30 min at room temperature. The mixture was partitioned, and the aqueous layer was extracted with CH2Cl2 (100 mL × 3). The combined organic layers were washed with H2O (100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was used for the next reaction without purification. A part of the crude product was purified by preparative thin-layer chromatography for analytical purposes.[α]27D + 43.4 (c = 1.06, CHCl3). IR (film) νmax 3672, 3301, 2987, 1749, 1374, 1216, 1166, 1071, 1023, 874. 1H-NMR (400 MHz, CDCl3) δ 1.31 (3H, s, acetonide), 1.33 (3H, s, acetonide), 1.43 (3H, s, acetonide), 1.52 (3H, s, acetonide), 1.58 (3H, d, J = 6.5 Hz, CHCH3), 3.93 (1H, dd, J = 8, 6.5 Hz, CHHCHCH), 4.04–4.19 (3H, m), 4.51 (1H, d, J = 3.5 Hz, OCHCHC), 4.96 (1H, q, J = 6.5 Hz, CHCH3), 5.82 (1H, d, J = 3.5 Hz, OCHCHC), 6.98 (1H, brs, NH). 13C-NMR (100 MHz, CDCl3) δ 16.4, 24.9, 26.1, 26.2, 26.5, 68.0, 70.1, 72.7, 76.1, 79.7, 87.2, 103.2, 109.9, 112.8, 159.0. HRMS (ESI) for C15H23NNaO7 (M+Na+), calcd 352.1372, found 352.1378.
N-Benzylcarbamate 15
The crude product 14 (10.5 g) was dissolved in dry THF (300 mL), and the solution was cooled to 0 °C under nitrogen. NaH (50–72% w/w, in mineral oil, 1.72 g, ca. 43.1 mmol), BnBr (4.10 mL, 34.4 mmol) and n-Bu4NI (5.30 g, 14.4 mmol) were successively added and stirring was continued for 14 h at room temperature. The mixture was quenched by addition of saturated aqueous NH4Cl (200 mL) and stirred for 30 min at room temperature. After H2O (100 mL) was added, the mixture was extracted with Et2O (100 mL × 3). The combined organic layers were washed with saturated aqueous NH4Cl (100 mL), H2O (100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 250 g) eluting with hexane–AcOEt (3:1) to give N-benzylcarbamate 15 (12.0 g, 99% in 2 steps) as a colorless oil.
[α]27D + 41.2 (c = 0.915, CHCl3). IR (film) νmax 2987, 1753, 1374, 1213, 1071, 1026, 872. 1H-NMR (400 MHz, CDCl3) δ 1.11 (3H, s, acetonide), 1.35 (3H, s, acetonide), 1.45 (3H, s, acetonide), 1.48 (3H, s, acetonide), 1.58 (3H, d, J = 6.5 Hz, CHCH3), 3.92 (1H, dd, J = 8.5, 6.5 Hz, CHHCHCH), 4.03 (1H, ddd, J = 8.5, 6.5, 6 Hz, CHHCHCH), 4.09 (1H, d, J = 16 Hz, PhCHH), 4.18 (1H, d, J = 8.5 Hz, CHHCHCH), 4.21 (1H, dd, J = 8.5, 6 Hz, CHHCHCH), 4.23 (1H, d, J = 4 Hz, OCHCHC), 4.81 (1H, q, J = 6.5 Hz, CHCH3), 5.04 (1H, d, J = 16 Hz, PhCHH), 5.69 (1H, d, J = 4 Hz, OCHCHC), 7.27–7.39 (5H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 16.5, 24.9, 25.8, 26.1, 26.3, 46.5, 68.5, 72.1, 73.4, 73.7, 81.2, 84.8, 103.4, 110.2, 112.5, 127.2, 128.0, 128.9, 137.8, 158.1. Elemental Analysis Calcdd for C22H29NO7: C, 62.99; H, 6.97; N, 3.34. found: C, 63.01; H, 6.90; N, 3.21%.
Hydrolysis of acetonide 15
A solution of N-benzylcarbamate 15 (12.0 g, 28.6 mmol) in 50% aqueous AcOH (300 mL) was stirred for 20 h at room temperature. The mixture was concentrated to dryness in vacuo. The crude product (12.2 g) was used for the next reaction without purification.A part of the crude product was purified by preparative thin-layer chromatography for analytical purposes.
IR (film) νmax 3412, 2936, 1992, 1734, 1409, 1295, 1215, 1098, 1073, 1025, 984, 872. 1H-NMR (400 MHz, CDCl3) δ 1.10 (3H, s, acetonide), 1.47 (3H, s, acetonide), 1.60 (3H, d, J = 6.5 Hz, CHCH3), 3.69 (1H, dd, J = 11, 6.5 Hz, CHHCHCH), 3.79 (1H, ddd, J = 9.5, 6.5 3 Hz, CHHCHCH), 3.92 (1H, dd, J = 11, 3 Hz, CHHCHCH), 4.09 (1H, d, J = 16 Hz, PhCHH), 4.21 (1H, d, J = 4 Hz, OCHCHC), 4.26 (1H, d, J = 9.5 Hz, CHHCHCH), 4.84 (1H, q, J = 6.5 Hz, CHCH3), 5.05 (1H, d, J = 16 Hz, PhCHH), 5.67 (1H, d, J = 4 Hz, OCHCHC), 7.27–7.38 (5H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 16.5, 25.9, 26.3, 46.5, 65.2, 69.3, 73.7, 74.3, 79.0, 84.7, 103.2, 112.4, 127.1, 128.0, 128.9, 137.7, 159.0. HRMS (ESI) for C19H26NO7 (M+H+), calcd 380.1709, found 380.1711.
Dimesylate
The crude product (12.2 g) was dissolved in dry CH2Cl2 (300 mL), and the solution was cooled to 0 °C under nitrogen. Et3N (12.0 mL, 85.9 mmol) and MsCl (5.50 mL, 71.6 mmol) were successively added and stirring was continued for 1 h at room temperature. The mixture was quenched by addition of saturated aqueous NH4Cl (200 mL) and stirred for 30 min at room temperature. After H2O (100 mL) was added, the mixture was extracted with Et2O (400 mL × 1, 100 mL × 2). The combined organic layers were washed with saturated aqueous NH4Cl (100 mL), H2O (100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The crude product (16.27 g) was used for the next reaction without purification. A part of the crude product was purified by preparative thin-layer chromatography for analytical purposes.IR (film) νmax 2985, 1753, 1358, 1178, 1094, 1068, 1028, 942. 1H-NMR (400 MHz, CDCl3) δ 1.14 (3H, s, acetonide), 1.49 (3H, s, acetonide), 1.55 (3H, d, J = 6.5 Hz, CHCH3), 3.09 (3H, s, Ms), 3.22 (3H, s, Ms), 4.14 (1H, d, J = 16 Hz, PhCHH), 4.29 (1H, d, J = 4 Hz, OCHCHC), 4.51 (1H, dd, J = 12, 2.5 Hz, CHHCHCH), 4.57 (1H, d, J = 9.5 Hz, CHHCHCH), 4.74 (1H, dd, J = 12, 2 Hz, CHHCHCH), 4.79 (1H, ddd, J = 9.5, 2.5, 2 Hz, CHcHdCHCH), 4.85 (1H, q, J = 6.5 Hz, CHCH3), 5.03 (1H, d, J = 16 Hz, PhCHaHb), 5.74 (1H, d, J = 4 Hz, OCHCHC), 7.30–7.41 (5H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 16.4, 26.0, 26.4, 37.4, 39.2, 46.6, 68.5, 72.9, 73.0, 73.7, 75.4, 85.3, 103.0, 113.2, 127.4, 128.3, 129.0, 137.1, 157.4. Elemental Analysis Calcdd for C21H29NO11S2: C, 47.09; H, 5.46; N, 2.62. found: C, 47.11; H, 5.38; N, 2.53%.
Vinyl compound 16
To a solution of the crude product (16.27 g) in DMF (300 mL) were added Zn (4.31 g, 65.9 mmol) and NaI (4.64 g, 34.4 mmol), and stirring was continued for 10 h at 120 °C under nitrogen. After cooling to room temperature, the reaction mixture was filtered through a pad of Super-Cel, and the precipitate was rinsed with Et2O (200 mL). The combined filtrate was washed with saturated aqueous NaHCO3 (100 mL) and saturated aqueous NH4Cl (100 mL). The combined aqueous layers were extracted with Et2O (100 mL × 2). The combined organic layers were washed with H2O (100 mL) and brine (100 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 100 g) eluting with hexane–AcOEt (2:1) to give 16 (9.15 g, 92% in 3 steps) as a colorless oil.
[α]25D + 48.8 (c = 1.10, CHCl3). IR (film) νmax 2988, 1756, 1397, 1191, 1068, 1028, 872. 1H-NMR (400 MHz, CDCl3) δ 1.10 (3H, s, acetonide), 1.48 (3H, s, acetonide), 1.50 (3H, d, J = 6.5 Hz, CHCH3), 4.15 (1H, d, J = 16 Hz, PhCHH), 4.18 (1H, d, J = 4 Hz, OCHCHC), 4.75 (1H, d, J = 5.5 Hz, CHHCHCH), 4.81 (1H, q, J = 6.5 Hz, CHCH3), 4.97 (1H, d, J = 16 Hz, PhCHH), 5.50 (1H, brd, J = 11 Hz, CHHCHCH), 5.64 (1H, d, J = 4 Hz, OCHCHC), 5.65 (1H, brd, J = 17 Hz, CHHCHCH), 5.86 (1H, ddd, J = 17, 11, 5.5 Hz, CHHCHCH), 7.27–7.38 (5H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 16.7, 25.7, 26.2, 46.4, 73.1, 73.3, 79.8, 84.3, 103.5, 112.3, 121.5, 127.4, 127.9, 128.8, 129.8, 137.7, 158.0. Elemental Analysis Calcdd for C19H23NO5: C, 66.07; H, 6.71; N, 4.06. found: C, 65.90; H, 6.62; N, 4.28%.
Hemiacetal 17 and its trifluoroacetate 18
A solution of 16 (3.09 g, 8.94 mmol) in TFA (72 mL) and H2O (18 mL) was stirred for 24 h at room temperature. The mixture was concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 50 g) eluting with hexane–AcOEt (2:1) to give diol 17 (1.76 g, 65%) as a white solid and trifluoroacetate 18 (1.03 g, 29%) as a pale red oil, and 0.13 g of starting material 16 (4%) were recovered. A solution of the trifluoroacetate 18 (1.03g, 2.57 mmol) and K2CO3 (300 mg) in MeOH (30 mL) was stirred for 2 h at room temperature. After addition of 1 N aqueous HCl (20 mL) and H2O (50 mL), the mixture was extracted with EtOAc (30 mL × 3). The combined organic layers were washed with saturated aqueous NH4Cl (20 mL), H2O (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 30 g) eluting with hexane–AcOEt (2:1) to give diol 17 (0.50 g, 62% from trifluoroacetate 18). Thus, 2.26 g (7.40 mmol) of diol 17 were obtained from 3.09 g of 16 in 83% yield.
17; 1H-NMR (400 MHz, CD3OD) δ 1.49 (2/3×3H, d, J = 6.5 Hz, CHCH3), 1.51 (1/3×3H, d, J = 6.5 Hz, CHCH3), 3.97 (1/3×1H, d, J = 5 Hz, OCHCHC), 4.04 (2/3×1H, d, J = 3 Hz, OCHCHC), 4.28 (1/3×1H, d, J = 16.5 Hz, PhCHH), 4.61 (2/3×1H, d, J = 16.5 Hz, PhCHH), 4.61 (2/3×1H, brd, J = 5.5 Hz, CHHCHCH), 4.69 (2/3×1H, d, J = 16.5 Hz, PhCHH), 4.78 (1/3×1H, brd, J = 5.5 Hz, CHHCHCH), 4.79 (1/3×1H, d, J = 16.5 Hz, PhCHH), 4.83 (2/3×1H, q, J = 6.5 Hz, CHCH3), 4.89 (1/3×1H, q, J = 6.5 Hz, CHCH3), 5.07 (2/3×1H, d, J = 3 Hz, OCHCHC), 5.32 (1/3×1H, d, J = 5 Hz, OCHCHC), 5.38 (2/3×1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.42 (1/3×1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.57 (2/3×1H, dt, J = 17, 1.5 Hz, CHHCHCH), 5.58 (1/3×1H, dt, J = 17, 1.5 Hz, CHHCHCH), 5.87 (1/3×1H, ddd, J = 17, 10.5, 5.5 Hz, CHHCHCH), 5.88 (2/3×1H, ddd, J = 17, 10.5, 5.5 Hz, CHHCHCH), 7.19–7.39 (5H, m, Ar). Elemental Analysis Calcd for C16H19NO5: C, 62.94; H, 6.27; N, 4.59. found: C, 62.94; H, 6.28; N, 4.59%.
18; IR (film) νmax 3199, 1721, 1415, 1194, 1099, 1073, 1004. 1H-NMR (300 MHz, CDCl3) δ 1.48 (1/3×3H, d, J = 6.5 Hz, CHCH3), 1.55 (2/3×3H, d, J = 6.5 Hz, CHCH3), 4.06 (2/3×1H, d, J = 16 Hz, PhCHH), 4.08 (1/3×1H, d, J = 16 Hz, PhCHH), 4.37 (2/3×1H, d, J = 4.5 Hz, OCHCHC), 4.52 (1/3×1H, d, J = 4 Hz, OCHCHC), 4.84 (1/3×1H, brd, J = 5.5 Hz, CHHCHCH), 4.86 (1/3×1H, q, J = 6.5 Hz, CHCH3), 4.88 (2/3×1H, q, J = 6.5 Hz, CHCH3), 4.99 (1H, d, J = 16 Hz, PhCHH), 5.02 (2/3×1H, brd, J = 5.5 Hz, CHHCHCH), 5.55 (2/3×1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.56 (1/3*1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.68 (1H, dt, J = 16, 1.5 Hz, CHHCHCH), 5.70 (2/3×1H, d, J = 4.5 Hz, OCHCHC), 5.84 (1/3×1H, ddd, J = 16, 10.5, 5.5 Hz, CHHCHCH), and (1/3×1H, d, J = 4 Hz, OCHCHC), 5.86 (2/3×1H, ddd, J = 16, 10.5, 5.5 Hz, CHHCHCH), 7.24–7.42 (5H, m, Ar). 19F-NMR (400 MHz, CDCl3) δ − 86.4 (1/3×3F, s, COCF3), −84.1 (2/3×3F, s, COCF3).
Aldehyde 20
To a solution of diol 17 (2.05 g, 6.71 mmol) in THF (35 mL) and H2O (35 mL) was added NaIO4 (2.87 g, 13.4 mmol), and stirring was continued for 1 h at room temperature. After H2O (30 mL) was added, the mixture was extracted with AcOEt (40 mL × 3). The combined organic layers were washed with H2O (40 mL) and brine (40 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The crude product 20 (2.02 g) was used for the next reaction without purification.
1H-NMR (300 MHz, CDCl3) δ 1.50 (3H, d, J = 7 Hz, CHCH3), 4.38 (1H, d, J = 15 Hz, PhCHH), 4.46 (1H, q, J = 7 Hz, CHCH3), 5.03 (1H, d, J = 15 Hz, PhCHH), 5.55 (1H, dt, J = 10.5, 1 Hz, CHHCHCH), 5.57 (1H, dt, J = 17, 1 Hz, CHHCHCH), 5.87 (1H, ddd, J = 17, 10.5, 6 Hz, CHHCHCH), 6.07 (1H, q, J = 6, 1 Hz, CHHCHCH), 7.28–7.42 (5H, m, Ar), 8.13 (1H, d, J = 1 Hz, OCHO), 8.94 (1H, s, CHO).
Propargyl alcohol 21 and 22
To a solution of trimethylsilylacetylene (4.0 mL, 28 mmol) in dry THF (50 mL) at 0 °C was added n-BuLi (1.65 M in THF 16.3 mL, 26.8 mmol) and stirring was continued for 30 min under nitrogen. To the solution was added the crude product 20 (2.02 g) in dry THF (25 mL) at −78 °C and stirring was continued for 25 min. The mixture was allowed to warm up to room temperature over 15 min. After stirring for an additional 30 min at room temperature, the mixture was quenched by addition of ice-cooled saturated aqueous NH4Cl (30 mL) and then stirred for 20 min. After H2O (30 mL) was added, the mixture was extracted with Et2O (40 mL × 3). The combined organic layers were washed with saturated aqueous NH4Cl (40 mL), H2O (40 mL) and brine (40 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 200 g) eluting with hexane–AcOEt (4:1 → 3:1 → 2:1) to give enyne 21 (1.53 g, 61% in 2 steps) as a white solid, and its diastereomer 22 (0.52 g, 21% in 2 steps) as a colorless oil.
21: [α]28D + 99.7 (c = 1.00, CHCl3). IR (film) νmax 3414, 2959, 1723, 1415, 1251, 1075, 845. 1H-NMR (400 MHz, CDCl3) δ 0.13 (9H, s, TMS), 1.52 (3H, d, J = 7 Hz, CHCH3), 4.58 (2H, s, PhCH2), 4.67 (1H, brd, J = 5 Hz, CHHCHCH), 4.93 (1H, brs, TMSC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH), 4.97 (1H, q, J = 7 Hz, CHCH3), 5.36 (1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.52 (1H, dt, J = 17, 1.5 Hz, CHHCHCH), 5.98 (1H, ddd, J = 17, 10.5, 5 Hz, CHHCHCH), 7.21–7.31 (3H, m, Ar) , 7.37–7.43 (2H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ − 0.4, 15.9, 46.6, 65.0, 69.9, 72.4, 75.8, 95.1, 101.7, 119.6, 127.5, 128.4, 128.7, 135.0, 137.9, 158.7. Elemental Analysis Calcdd for C20H27NO4Si: C, 64.31; H, 7.29; N, 3.75. found: C, 64.32; H, 7.05; N, 3.77%.
CCH), 4.97 (1H, q, J = 7 Hz, CHCH3), 5.36 (1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.52 (1H, dt, J = 17, 1.5 Hz, CHHCHCH), 5.98 (1H, ddd, J = 17, 10.5, 5 Hz, CHHCHCH), 7.21–7.31 (3H, m, Ar) , 7.37–7.43 (2H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ − 0.4, 15.9, 46.6, 65.0, 69.9, 72.4, 75.8, 95.1, 101.7, 119.6, 127.5, 128.4, 128.7, 135.0, 137.9, 158.7. Elemental Analysis Calcdd for C20H27NO4Si: C, 64.31; H, 7.29; N, 3.75. found: C, 64.32; H, 7.05; N, 3.77%.
22: [α]27D + 34.4 (c = 1.04, CHCl3). IR (film) νmax 3543, 3403, 2959, 1726, 1415, 1251, 1071, 845. 1H-NMR (400 MHz, CDCl3) δ 0.16 (9H, s, TMS), 1.19 (1H, d, J = 4.5 Hz, TMSCCCHOH), 1.53 (3H, d, J = 7 Hz, CHCH3), 2.09 (1H, d, J = 4 Hz, CHHCHCHOH), 4.49 (1H, d, J = 15.5 Hz, PhCHH), 4.73 (1H, brd, J = 5 Hz, CHHCHCH), 4.83 (1H, q, J = 7 Hz, CHCH3), 4.88 (1H, d, J = 15.5 Hz, PhCHH), 4.89 (1H, d, J = 4.5 Hz, TMSCCCH), 5.45 (1H, dt, J = 10.5, 1.5 Hz, CHHCHCH), 5.61 (1H, dt, J = 17, 1.5 Hz, CHHCHCH), 6.05 (1H, ddd, J = 17, 10.5, 5 Hz, CHHCHCH), 7.28–7.39 (3H, m, Ar) , 7.42–7.47 (2H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ − 0.4, 15.8, 46.0, 60.7, 71.4, 72.9, 73.5, 92.5, 101.2, 119.3, 128.3, 128.4, 129.4, 135.4, 138.2, 158.5. Elemental Analysis Calcdd for C20H27NO4Si: C, 64.31; H, 7.29; N, 3.75. found: C, 64.33; H, 7.13; N, 3.66%.
Preparation of acetylene–Co2(CO)6 complex
To a solution of enyne 21 (100 mg, 0.268 mmol) in dry CH2Cl2 (3 mL) was added Co2(CO)8 (137 mg, 0.402 mmol). After stirring for 3 h at room temperature, the mixture was concentrated to dryness in vacuo. The residue was purified by column chromatography (neutral silica gel 10 g, hexane–AcOEt = 4:1) to give the acetylene–cobalt complex (155 mg, 88%) as a brown oil.
1H-NMR (400 MHz, CDCl3) δ 0.37 (9H, s, TMS), 1.08 (1H, brd, J = 7 Hz, CHHCHCHOH), 1.47 (3H, d, J = 6.5 Hz, CHCH3), 3.18 (1H, d, J = 6 Hz, CCH(OH)C), 4.00 (1H, d, J = 15.5 Hz, PhHH), 4.58 (1H, brt, J = 6 Hz, CHHCHCHOH), 4.83 (1H, brq, J = 6.5 Hz, CHCH3), 4.97 (1H, brd, J = 17 Hz, CHHCHCHOH), 5.01–5.17 (2H, m, CHHCHCHOH, PhHH), 5.70 (1H, d, J = 6 Hz, CCH(OH)C), 5.78 (1H, m, CHHCHCH), 7.30–7.38 (3H, br, Ar) , 7.45–7.53 (2H, br, Ar). HRMS (ESI) for C26H27Co2NNaO10Si (M+Na+), calcd 681.9966, found 681.9977.
Cyclopentenones 23 and 24
A solution of acetylene–cobalt complex (100 mg, 0.152 mmol) in dry CH3CN (2 mL) was stirred for 1 h at 70 °C. After cooling to room temperature, the reaction mixture was filtered through a pad of Super-Cel, and the precipitate was rinsed with CH3CN (10 mL). The combined filtrate was concentrated to dryness in vacuo. The residue was purified by column chromatography (neutral silica gel 20 g) eluting with hexane–acetone (4:1) to give cycloadduct 23 (27.3 mg, 45%) as a white solid and its diastereomer 24 (14.2 mg, 23%) as a colorless oil.
23: [α]27D + 69.2 (c = 0.38, acetone). IR (film) νmax 3361, 1734, 1684, 1407, 1247, 1148, 844. 1H-NMR (400 MHz, CDCl3) δ 0.27 (9H, s, TMS), 1.58 (3H, d, J = 6 Hz, CHCH3), 2.16 (1H, dd, J = 18, 4 Hz, CHCHHCO), 2.26 (1H, dd, J = 18, 6.5 Hz, CHCHHCO), 3.42 (1H, m, CHCHHCO), 4.03 (1H, dd, J = 4 Hz, CCHOH), 4.10 (1H, d, J = 17 Hz, PhCHH), 4.56 (1H, q, J = 6 Hz, CHCH3), 4.97 (1H, brs, CHCHOH), 5.22 (1H, d, J = 17 Hz, PhCHH), 7.23–7.31 (3H, m, Ar) , 7.31–7.38 (2H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ − 1.3, 17.8, 37.3, 47.7, 50.4, 71.1, 75.6, 78.6, 80.4, 126.6, 127.7, 129.0, 138.1, 142.8, 160.1, 189.1, 214.3. Elemental Analysis Calcd for C20H27NO4Si: C, 62.82; H, 6.78; N, 3.49. found: C, 62.81; H, 6.59; N, 3.41%.
24: [α]22D − 161 (c = 0.30, CDCl3). IR (film) νmax 3361, 1734, 1684, 1407, 1247, 1148, 844. 1H-NMR (400 MHz, CDCl3) δ 0.28 (9H, s, TMS), 0.71 (1H, brs, CHCHOH), 1.48 (3H, d, J = 6.5 Hz, CHCH3), 2.20 (1H, dd, J = 18, 3.5 Hz, CHCHHCO), 2.61 (1H, dd, J = 18, 6.5 Hz, CHCHHCO), 2.83 (1H, ddd, J = 12, 6.5, 3.5 Hz, CHCHHCO), 4.06 (1H, d, J = 12 Hz, CHCHOH), 4.43 (1H, q, J = 6.5 Hz, CHCH3), 4.58 (1H, d, J = 16 Hz, PhCHH), 5.02 (1H, brs, CCHOH), 5.09 (1H, d, J = 16 Hz, PhCHH), 5.48 (1H, brd, J = 4 Hz, CCHOH), 7.30–7.45 (5H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ − 1.3, 17.8, 42.3, 46.2, 48.3, 73.2, 75.2, 76.1, 80.3, 127.4, 128.4, 129.5, 138.2, 144.9, 159.3, 186.7, 211.5. HRMS (ESI) for C21H28NO5Si (M+H+), calcd 402.1737, found 402.1740.
25: [α]26D + 26.7 (c = 0.55, CHCl3). IR (NaCl film) νmax 2956, 1755, 1705, 1621, 1497, 1386, 1212, 1086, 976, 887. 1H-NMR (400 MHz, CDCl3) δ 0.21 (9H, s, TMS), 1.37 (3H, d, J = 6 Hz, CHCH3), 1.77 (1H, dd, J = 17.5, 4.5 Hz, CHCHHCO), 1.97 (3H, s, Ac), 2.13 (3H, s, Ac), 2.33 (1H, dd, J = 17.5, 7 Hz, CHCHHCO), 3.54 (1H, m, CHCHH), 4.08 (1H, d, J = 17 Hz, PhCHH), 4.70 (1H, q, J = 6 Hz, CHCH3), 5.24 (1H, d, J = 5.5 Hz, CHCHOAc), 5.24 (1H, d, J = 17 Hz, PhCHH), 6.04 (1H, d, J = 1.5 Hz, CCH(OAc)C), 7.41–7.24 (5H, m, Ph). 13C-NMR (75 MHz, CDCl3) δ − 1.6, 17.5, 20.3, 20.4, 38.9, 47.2, 48.5, 73.4, 74.9, 76.8, 77.8, 126.2, 128.1, 129.2, 137.1, 144.7, 158.2, 168.9, 169.1, 181.7, 211.2. HRMS (FAB+) for C25H32NO7Si (M+H) calcd 486.1948, found 486.1930.
Desilylation of 21
To a solution of enyne 21 (832 mg, 2.23 mmol) in MeOH (60 mL) was added K2CO3 (462 mg, 3.34 mmol) and stirring was continued for 3 h at room temperature. The mixture was quenched by addition of 1 N aqueous HCl (5 mL) and stirred for 30 min at room temperature. After H2O (50 mL) was added, the mixture was extracted with AcOEt (30 mL × 3). The combined organic layers were washed with H2O (30 mL) and brine (30 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The crude product (672 mg) was used for the next reaction without purification.Dibenzyl ether 26
The crude product (672 mg) was dissolved in dry THF (60 mL), and the solution was cooled to 0 °C under nitrogen. BnBr (0.66 mL, 5.58 mmol), n-Bu4NI (823 mg, 2.23 mmol) and NaH (50–72% w/w, in mineral oil, 223 mg) were successively added and stirring was continued for 12 h at room temperature. The mixture was quenched by addition of saturated aqueous NH4Cl (20 mL) and stirred for 30 min at room temperature. After H2O (20 mL) was added, the mixture was extracted with Et2O (30 mL × 3). The combined organic layers were washed with saturated aqueous NH4Cl (30 mL), H2O (30 mL) and brine (30 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 50 g) eluting with hexane–AcOEt (4:1) to give enyne 26 (836 mg, 78% in 2 steps) as a colorless oil.
[α]26D + 30.4 (c = 1.10, CHCl3). IR (film) νmax 3282, 3031, 1750, 1403, 1069. 1H-NMR (400 MHz, CDCl3) δ 1.49 (3H, d, J = 7 Hz, CHCH3), 2.29 (1H, d, J = 2 Hz, CCHCCH), 4.10 (1H, d, J = 11 Hz, PhCHaHb), 4.23 (1H, d, J = 11 Hz, PhCHcHd), 4.30 (1H, d, J = 7 Hz, CHHCHCH), 4.47 (1H, d, J = 15.5 Hz, PhCHeHf), 4.56 (1H, d, J = 11 Hz, PhCHaHb), 4.70 (1H, d, J = 15.5 Hz, PhCHeHf), 4.72 (1H, d, J = 11 Hz, PhCHcHd), 4.89 (1H, d, J = 2 Hz, CCHCCH), 5.05 (1H, q, J = 7 Hz, CHCH3), 5.49 (1H, brd, J = 17.5 Hz, CHHCHCH), 5.52 (1H, brd, J = 10.5 Hz, CHHCHCH), 5.78 (1H, ddd, J = 17.5, 10.5, 7 Hz, CHHCHCH), 7.16–7.37 (15H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 15.6, 46.9, 70.4, 70.8, 71.3, 71.5, 75.6, 78.3, 79.0, 80.1, 121.9, 127.3, 127.6, 127.7, 127.8, 127.9, 128.0, 128.4 × 2, 129.4, 132.2, 137.0, 137.7, 137.8, 158.7. Elemental Analysis Calcd for C31H31NO4: C, 77.31; H, 6.49; N, 2.91. found: C, 77.32; H, 6.55; N, 2.82%.
Aldehyde 27
A solution of enyne 26 (197 mg, 0.409 mmol) in CH2Cl2 (40 mL) was cooled to −78 °C under argon. Ozone was passed into the solution until the color became blue, and the solution was stirred for 30 min at −78 °C. After the excess of ozone was purged with argon, PPh3 (161 mg, 0.613 mmol) was added at −78 °C. The mixture was allowed to warm up to room temperature and stirred for 1 h. The mixture was concentrated to dryness in vacuo. The residue was purified by column chromatography (silica gel 25 g) eluting with hexane–EtOAc (4:1) to give aldehyde 27 (167 mg, 0.345 mmol, 84%) as a colorless oil.
[α]27D − 64.6 (c = 1.14, CHCl3). IR (film) νmax 3279, 1749, 1402, 1089, 1070. 1H-NMR (400 MHz, CDCl3) δ 1.57 (3H, d, J = 6.5 Hz, CHCH3), 2.51 (1H, d, J = 1.5 Hz, CCHCCH), 4.07 (1H, d, J = 11 Hz, PhCHaHb), 4.13 (1H, d, J = 11 Hz, PhCHcHd), 4.17 (1H, d, J = 2 Hz, CHCHO), 4.36 (1H, d, J = 15.5 Hz, PhCHeHf), 4.42 (1H, d, J = 15.5 Hz, PhCHeHf), 4.44 (1H, d, J = 11 Hz, PhCHaHb), 4.63 (1H, brs, CCHCCH), 4.71 (1H, d, J = 11 Hz, PhCHcHd), 4.93 (1H, q, J = 6.5 Hz, CHCH3), 7.14–7.38 (15H, m, Ar), 9.60 (1H, d, J = 2 Hz, CHCHO). 13C-NMR (100 MHz, CDCl3) δ 16.6, 46.2, 70.2, 70.9, 71.3, 73.7, 75.8, 79.5, 83.2, 127.6, 128.0 × 2, 128.2, 128.3 × 2, 128.5, 128.6, 129.0, 136.3, 136.4, 137.3, 158.0, 198.6. Elemental Analysis Calcd for C30H29NO5: C, 74.52; H, 6.04; N, 2.90. found: C, 74.52; H, 6.03; N, 2.82%.
Nitrone 28
Aldehyde 27 (66.8 mg, 0.138 mmol) and N-PhNHOH (18.1 mg, 0.166 mmol) were dissolved in toluene (5 mL). The mixture was concentrated to dryness in vacuo over 30 min below room temperature. The residue was purified by column chromatography (neutral silica gel 5 g) eluting with hexane–AcOEt (3:1) to give nitrone 28 (68.0 mg, 86%) as a colorless oil. During the purification, purified 28 gradually underwent the Baldwin rearrangement to give a mixture of 28 and a small amount of aziridine 30.
IR (film) νmax 2925, 2360, 1750, 1397, 1067. 1H-NMR (300 MHz, CDCl3) δ 1.44 (3H, d, J = 7 Hz, CHCH3), 2.48 (1H, d, J = 2 Hz, CCHCCH), 4.28–4.42 (4H, m), 4.74 (1H, d, J = 15 Hz, PhCHaHb), 4.80 (1H, d, J = 11.5 Hz, PhCHaHb), 5.07 (1H, d, J = 2 Hz, CCHCCH), 5.09 (1H, q, J = 7 Hz, CHCH3), 5.42 (1H, d, J = 8.5 Hz, CHCHN), 6.89 (1H, d, J = 8.5 Hz, CHCHN), 7.12–7.46 (15H, m, Ar). HRMS (ESI) for C36H34N2NaO5 (M+Na), calcd 597.2365, found 597.2370.
Aziridine aldehyde 30
A solution of nitrone 28 (42.0 mg, 73.1 μmol) in toluene (10 mL) was heated to reflux temperature for 30 min under nitrogen. The mixture was concentrated to dryness in vacuo. The residue was purified by column chromatography (neutral silica gel 5 g) eluting with hexane–AcOEt (4:1) to give aldehyde 30 (33.1 mg, 79%) as a colorless oil.
[α]27D + 50.8 (c = 1.78, CHCl3). IR (film) νmax 3031, 1749, 1703, 1595, 1492, 1454, 1389, 1089, 1072. 1H-NMR (400 MHz, CDCl3) δ 1.46 (3H, d, J = 6 Hz, CHCH3), 3.07 (1H, s, NCHCH), 3.83 (1H, s, NCCH), 4.10 (1H, d, J = 12 Hz, PhCHaHb), 4.38 (1H, q, J = 6 Hz, CHCH3), 4.39 (1H, d, J = 12 Hz, PhCHaHb), 4.70 (1H, d, J = 16 Hz, PhCHcHd), 4.73 (1H, d, J = 11.5 Hz, PhCHeHf), 4.76 (1H, s, NCHCH), 4.84 (1H, d, J = 11.5 Hz, PhCHeHf), 4.94 (1H, d, J = 16 Hz, PhCHcHd), 6.63 (2H, brd, J = 7.5 Hz, Ar), 7.04 (2H, brt, J = 7.5 Hz, Ar), 7.07–7.16 (4H, m, Ar), 7.20–7.44 (14H, m, Ar), 8.82 (1H, s, CHO). 13C-NMR (100 MHz, CDCl3) δ 17.8, 47.1, 49.7, 58.9, 73.0, 73.8, 78.8, 80.1, 81.6, 120.0, 124.0, 127.1, 127.8, 127.9, 128.1, 128.4 × 2, 128.5, 128.6, 128.8, 129.7, 136.3, 137.3, 139.4, 145.1, 158.8, 194.8. Elemental Analysis Calcd for C36H34N2O5: C, 75.24; H, 5.96; N, 4.87. found: C, 75.24; H, 5.78; N, 4.93%.
Reduction of aziridine aldehyde 30
To a solution of aldehyde 30 (49.6 mg, 86.3 μmol) in MeOH (6 mL) was added NaBH4 (6.5 mg, 0.17 mmol) and stirring was continued for 20 min at room temperature. The mixture was quenched by addition of saturated aqueous NH4Cl (10 mL) and stirred for 30 min at room temperature. After H2O (5 mL) was added, the mixture was extracted with AcOEt (10 mL × 1, 5 mL × 2). The combined organic layers were washed with H2O (5 mL) and brine (5 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (neutral silica gel 5 g) eluting with hexane–AcOEt (3:2) to give the product (42.8 mg, 86%) as a colorless oil.
[α]27D + 12.4 (c = 1.40, CHCl3). IR (film) νmax 3662, 3183, 3057, 1733, 1595, 1396, 1262, 1070, 894. 1H-NMR (400 MHz, CDCl3) δ 0.96 (1H, brs, CHHOH), 1.49 (3H, d, J = 6 Hz, CHCH3), 2.55 (1H, s, NCHCH), 3.40 (1H, brd, J = 12.5 Hz, CHHOH), 3.60 (1H, dd, J = 12.5, 5.5 Hz, CHHOH), 3.80 (1H, s, NCCH), 4.22 (1H, d, J = 11.5 Hz, PhCHaHb), 4.29 (1H, s, NCHCH), 4.32 (1H, d, J = 11.5 Hz, PhCHaHb), 4.46 (1H, q, J = 6 Hz, CHCH3), 4.66 (1H, d, J = 11.5 Hz, PhCHcHd), 4.83 (1H, d, J = 11.5 Hz, PhCHcHd), 4.86 (1H, d, J = 15.5 Hz, PhCHeHf), 4.95 (1H, d, J = 15.5 Hz, PhCHeHf), 6.67 (2H, brd, J = 7.5 Hz, Ar), 6.99 (1H, brt, J = 7.5 Hz, Ar), 7.08–7.15 (4H, m, Ar), 7.20–7.45 (13H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 18.2, 45.3, 47.1, 54.0, 59.5, 73.1, 73.8, 76.8, 79.0, 80.6, 86.7, 120.1, 122.7, 126.9, 128.0, 128.1, 128.3 × 2, 128.4, 128.5 × 2, 128.6, 129.3, 136.6, 138.0, 139.7, 146.9, 159.0. HRMS (ESI) for C36H36N2O5Na (M+Na), calcd 599.2522, found 599.2511.
Mesylate 31
To a solution of the alcohol (41.2 mg, 71.4 μmol) in dry CH2Cl2 (5 mL) were added Et3N (25 μL, 0.179 mmol) and MsCl (12 μL, 0.155 mmol) at 0 °C under nitrogen, and stirring was continued for 1 h at room temperature. The mixture was quenched by addition of saturated aqueous NH4Cl (10 mL) and stirred for 30 min at room temperature. After H2O (5 mL) was added, the mixture was extracted with Et2O (10 mL × 1, 5 mL × 2). The combined organic layers were washed with H2O (5 mL) and brine (5 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by column chromatography (neutral silica gel 5 g) eluting with hexane–AcOEt (2:1) to give mesylate 31 (41.0 mg, 88%) as a colorless oil.
[α]27D + 21.1 (c = 1.06, CHCl3). IR (film) νmax 3030, 1744, 1494, 1359, 1175, 1086, 953, 815. 1H-NMR (400 MHz, CDCl3) δ 1.49 (3H, d, J = 6 Hz, CHCH3), 2.57 (3H, s, Ms), 2.63 (1H, s, NCHCH), 3.76 (1H, s, NCCH), 3.87 (1H, d, J = 11.5 Hz, CHHOMs), 4.09 (1H, d, J = 11.5 Hz, CHHOMs), 4.23 (1H, d, J = 11.5 Hz, PhCHaHb), 4.32 (1H, s, NCHCH), 4.34 (1H, d, J = 11.5 Hz, PhCHaHb), 4.47 (1H, q, J = 6 Hz, CHCH3), 4.64 (1H, d, J = 11.5 Hz, PhCHcHd), 4.82 (1H, d, J = 15.5 Hz, PhCHeHf), 4.88 (1H, d, J = 11.5 Hz, PhCHcHd), 4.99 (1H, d, J = 15.5 Hz, PhCHeHf), 6.66 (2H, brd, J = 7.5 Hz, Ar), 7.04 (1H, brt, J = 7.5 Hz, Ar), 7.07–7.14 (4H, m, Ar), 7.22–7.47 (13H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 18.2, 37.2, 45.7, 47.2, 51.0, 65.9, 73.1, 74.4, 76.3, 78.6, 80.6, 87.2, 120.4, 123.4, 127.0, 128.0, 128.1, 128.4 × 3, 128.6, 128.7 × 2, 129.5, 136.3, 137.8, 139.6, 145.6, 158.9. HRMS (ESI) for C36H36N2O5Na (M+Na), calcd 677.2297, found 677.2316.
Iodide 32
To a solution of mesylate 31 (38.2 mg, 58.4 μmol) in dry THF (5 mL) was added n-Bu4NI (32.3 mg, 87.5 μmol), and stirring was continued for 22 h at 60 °C under nitrogen. The mixture was concentrated to dryness in vacuo. The residue was purified by column chromatography (neutral silica gel 5 g) eluting with hexane–AcOEt (5:1) to give iodide 32 (29.0 mg, 73%) as a pale yellow oil.
[α]26D − 105 (c = 1.05, CHCl3). IR (film) νmax 3020, 1741, 1522, 1426, 1216, 929. 1H-NMR (400 MHz, CDCl3) δ 1.47 (3H, d, J = 6 Hz, CHCH3), 2.39 (1H, s, NCHCH), 2.56 (1H, d, J = 11 Hz, CHHI), 3.06 (1H, d, J = 11 Hz, CHHI), 3.76 (1H, s, NCCH), 4.30 (1H, d, J = 16 Hz, PhCHaHb), 4.33 (1H, d, J = 16 Hz, PhCHaHb), 4.52 (1H, q, J = 6 Hz, CHCH3), 4.59 (1H, d, J = 12 Hz, PhCHcHd), 4.62 (1H, s, NCHCH), 4.84 (1H, d, J = 15.5 Hz, PhCHeHf), 4.95 (1H, d, J = 12 Hz, PhCHcHd), 4.99 (1H, d, J = 15.5 Hz, PhCHeHf), 6.63 (2H, brd, J = 7.5 Hz, Ar), 7.03 (1H, brt, J = 7.5 Hz, Ar), 7.06–7.11 (2H, m, Ar), 7.14–7.19 (2H, m, Ar), 7.21–7.48 (13H, m, Ar). 13C-NMR (100 MHz, CDCl3) δ 1.4, 18.2, 47.4, 49.8, 54.4, 73.0, 75.0, 76.3, 79.0, 80.8, 87.5, 120.7, 123.2, 126.9, 128.1, 128.2, 128.3 × 2, 128.6 × 2, 128.7, 129.4, 136.5, 138.3, 139.9, 146.0, 159.0. HRMS (ESI) for C36H36IN2O5Na (M+Na), calcd 709.1539, found 709.1550.
exo-Olefin 33
To a solution of iodide 32 (20.5 mg, 29.9 μmol) in dry CH2Cl2 (3 mL) were added n-Bu4NI (16.5 mg, 44.8 μmol) and BF3·OEt2 (40 μL, 32.4 μmol) and stirring was continued for 20 min at 0 °C under nitrogen. The mixture was quenched by addition of crushed ice and the mixture was allowed to warm up to room temperature and stirring was continued for 10 min. After saturated aqueous NaHCO3 (10 mL) was added, the mixture was extracted with CH2Cl2 (5 mL × 3). The combined organic layers were washed with H2O (5 mL) and brine (5 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by preparative thin-layer chromatography (hexane–AcOEt 3:1) to give allylamine 33 (13.2 mg, 79%) as a white solid.
[α]28D − 7.3 (c = 0.59, CHCl3). mp: 142–146 °C (Hexane–AcOEt). IR (film) νmax 1747, 1602, 1497, 1398, 1216, 1087. 1H-NMR (400 MHz, C6D6) δ 1.51 (3H, d, J = 6.5 Hz, CHCH3), 3.38 (1H, s, CHHCCHC), 3.80 (1H, d, J = 11.5 Hz, PhCHaHb), 3.86 (1H, q, J = 6.5 Hz, CHCH3), 4.02 (1H, d, J = 11.5 Hz, PhCHcHd), 4.12 (1H, d, J = 11.5 Hz, PhCHaHb), 4.14 (1H, brd, J = 7 Hz, NHCHCH), 4.28 (1H, d, J = 7 Hz, NHCHCH), 4.34 (1H, d, J = 11.5 Hz, PhCHcHd), 4.67 (1H, d, J = 16 Hz, PhCHeHf), 4.70 (1H, d, J = 2 Hz, CHHCCHC), 4.77 (1H, d, J = 16 Hz, PhCHeHf), 5.01 (1H, brs, CHHCCHC), 6.57 (2H, brd, J = 8 Hz, Ar), 6.76 (1H, brt, J = 7.5 Hz, Ar), 6.92–6.96 (3H, m, Ar), 6.99–7.19 (12H, m, Ar), 7.35 (2H, brd, J = 7.5 Hz, Ar). 13C-NMR (100 MHz, CDCl3) δ 17.4, 46.2, 59.6, 69.8, 71.2, 73.2, 78.1, 85.6, 85.9, 114.1, 117.0, 118.6, 126.8, 126.9, 127.5, 127.6, 128.1 × 2, 128.2, 128.4, 128.6, 129.4, 136.8, 137.7, 138.7, 145.3, 146.6, 158.4. HRMS (ESI) for C36H38N2O6Na (M+Na+), calcd 617.2628, found 617.2610.
Diol 34
To a solution of allylamine 33 (10.6 mg, 18.9 μmol) in THF (0.5 mL) and H2O (0.5 mL) were added NMO (4.4 mg, 38 μmol) and 4% aqueous OsO4 (12 μL, 1.9 μmol), and stirring was continued for 24 h at room temperature. The mixture was quenched by addition of saturated aqueous Na2S2O3 (3 mL) and stirred for 30 min at room temperature. After H2O (2 mL) was added, the mixture was extracted with AcOEt (3 mL × 3). The combined organic layers were washed with H2O (3 mL) and brine (3 mL), dried over Na2SO4, and concentrated to dryness in vacuo. The residue was purified by preparative thin-layer chromatography (hexane–AcOEt = 1:1) to give diol 34 (6.6 mg, 59%) as a colorless oil.
[α]23D − 3.3 (c = 0.28, CHCl3). IR (film) νmax 3734, 3365, 1718, 1603, 1497, 1418, 1099, 1028. 1H-NMR (400 MHz, C6D6) δ 1.58 (3H, d, J = 6.5 Hz, CHCH3), 2.88 (1H, brs, t-OH), 3.48 (1H, d, J = 11 Hz, CHaHbOH), 3.65 (1H, s, CCHC), 3.71 (1H, d, J = 11.5 Hz, PhCHcHd), 3.76 (1H, d, J = 11 Hz, CHaHbOH), 3.86 (1H, d, J = 9.5 Hz, NHCHCH), 4.00 (1H, d, J = 9.5 Hz, NHCHCH), 4.18 (1H, d, J = 11.5 Hz, PhCHcHd), 4.36 (1H, d, J = 10.5 Hz, PhCHeHf), 4.41 (1H, d, J = 10.5 Hz, PhCHeHf), 4.59 (1H, d, J = 16.5 Hz, PhCHgHh), 4.79 (1H, q, J = 6.5 Hz, CHCH3), 5.04 (1H, d, J = 16.5 Hz, PhCHgHh), 6.63 (2H, brd, J = 8, Hz, Ar), 6.72 (1H, brt, J = 7.5 Hz, Ar), 6.85 (2H, brdd, J = 8, 1.5 Hz, Ar), 6.93–7.24 (13H, m, Ar), 7.40 (2H, brd, J = 7.5 Hz, Ar). 13C-NMR (100 MHz, CDCl3) δ 17.1, 46.4, 63.4, 65.4, 70.2, 73.6, 75.8, 79.2, 79.6, 83.5, 88.8, 113.7, 118.8, 127.2, 127.3 × 2, 127.6, 128.2, 128.5, 128.6, 128.7, 128.9, 129.5, 136.5, 137.4, 137.9, 147.0, 158.8. HRMS (ESI) for C36H37N2O4 (M+H+), calcd 561.2753, found 561.2735.
Acknowledgements
We are grateful to Prof. Kin-ichi Tadano (Keio University, Japan) for valuable discussions. We thank Dr Daisuke Urabe for his early studies of the Overman rearrangement of compound 6. Elemental analyses and measurement of 400 MHz NMR spectra were performed by Mr. S. Kitamura and K. Koga (analytical laboratory of this school), whom we thank. Financial support was provided by a Grant in Aid for Scientific Research and a G-COE grant from MEXT, PRESTO from JST, the Nagase Foundation, and the Sumitomo Foundation.References
- (a) B. K. Bhuyan, A. Dietz and C. G. Smith, Antimicrobial Agents Chemother., 1962, 184–190 CAS; (b) A. D. Argoudelis, H. K. Jahnke and J. A. Fox, Antimicrobial Agents Chemother., 1962, 191–197 CAS; (c) B. K. Bhuyan, Appl. Microbiol., 1962, 10, 302–304 CAS.
- P. F. Wiley, H. K. Jahnke, F. MacKellar, R. B. Kelly and A. D. Argoudelis, J. Org. Chem., 1970, 35, 1420–1425 CrossRef CAS.
- (a) D. J. Duchamp, American Crystallographic Association Winter Meeting, Albuquerque, NM 1972, Abstracts, p. 23 Search PubMed; (b) For assignment of 13C NMR of pactamycin, see: D. D. Weller, A. Haber, K. L. Rinehart, Jr. and P. F. Wiley, J. Antibiot., 1978, 31, 997–1006 CrossRef CAS.
- E. S. Adama and K. L. Rinehart, J. Antibiot., 1994, 47, 1456–1465 CrossRef.
- T. A. Beerman, I. H. Goldberg, L. S. Kappen, R. Poon and H. Suzuki, Adv. Enzyme Regul., 1976, 14, 207–225 CrossRef CAS.
- L. B. Cohen, I. H. Goldberg and A. E. Herner, Biochemistry, 1969, 8, 1327–1335 CrossRef CAS.
- (a) The crystal structure of the complex of pactamycin with a ribosomal 30S subunit was reported. D. E. Brodersen, W. M. Clemons, Jr., A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Cell, 2000, 103, 1143–1154 CrossRef CAS; (b) G. Dinos, D. N. Wilson, Y. Teraoka, W. Szaflarski, P. Fucini, D. Kalpaxis and K. H. Nierhaus, Mol. Cell, 2004, 13, 113–124 CrossRef CAS.
- D. D. Weller and K. L. Rinehart Jr., J. Am. Chem. Soc., 1978, 100, 6757–6760 CrossRef CAS.
- T. R. Hurley, T. A. Smitka, J. H. Wilton, R. H. Bunge, G. C. Hokanson and J. C. French, J. Antibiot., 1986, 39, 1086–1091 CrossRef CAS.
- M. Iwatsuki, A. Nishihara-Tsukashima, A. Ishiyama, M. Namatame, Y. Watanabe, S. Handasah, H. Pranamuda, B. Marwoto, A. Matsumoto, Y. Takahashi, K. Otoguro and S. Ōmura, J. Antibiot., 2012, 65, 169–171 CrossRef CAS.
- S. Knapp and Y. Yu, Org. Lett., 2007, 9, 1359–1362 CrossRef CAS.
- T. Tsujimoto, T. Nishikawa, T. Urabe and M. Isobe, Synlett, 2005, 433–436 CAS.
- S. Hanessian, R. R. Vakiti, S. Dorich, S. Banerjee, F. Lecomte, J. R. DelValle, J. Zhang and B. Deschênes-Simard, Angew. Chem., Int. Ed., 2011, 50, 3497–3500 CrossRef CAS.
- J. T. Malinowski, S. J. McCarver and J. S. Johnson, Org. Lett., 2012, 14, 2878–2881 CrossRef CAS.
- T. J. Haussener and R. E. Looper, Org. Lett., 2012, 14, 3632–3635 CrossRef CAS.
- For preliminary report, see ref. 12.
- The numbering used in this paper corresponds to that of pactamycin.
- For reviews of the Pauson–Khand reaction, see: (a) P. L. Pauson, Tetrahedron, 1985, 41, 5855–5860 CrossRef CAS; (b) N. E. Shore, Org. React., 1991, 40, 1–90 Search PubMed; (c) J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC.
- For reviews of the Overman rearrangement, see: (a) L. E. Overman, Acc. Chem. Res., 1980, 13, 218–224 CrossRef CAS; (b) L. E. Overaman and N. E. Carpenter, Org. React., 2005, 66, 1–107 Search PubMed.
- (a) K.-I. Tadano, Y. Idogaki, H. Yamada and T. Suami, Chem. Lett., 1985, 1925–1928 CrossRef CAS; (b) K.-I. Tadano, Y. Idogaki, H. Yamada and T. Suami, J. Org. Chem., 1987, 52, 1201–1210 CrossRef CAS; (c) K.-I. Tadano, J. Ishihara and S. Ogawa, J. Synth. Org. Chem. Jpn., 1991, 49, 327–339 CrossRef CAS(in Japanese).
- For reviews of the Ley oxidation, see: (a) W.P. Griffith and S. V. Ley, Aldrichmica, Acta, 1990, 23, 13–19 CAS; (b) S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639–666 CrossRef.
- (a) J. Gonda and M. Bednárikova, Tetrahedron Lett., 1997, 38, 5569–5572 CrossRef CAS; (b) T. Eguchi and K. Kakinuma, J. Synth. Org. Chem. Jpn., 1997, 55, 814–823 CrossRef CAS (in Japanese).
- T. Nishikawa, M. Asai, N. Ohyabu and M. Isobe, J. Org. Chem., 1998, 63, 188–192 CrossRef.
- (a) G. Cardillo, M. Orena and S. Sandri, J. Chem. Soc., Chem. Commun., 1983, 1489–1490 RSC; (b) G. Cardillo, in Houben-Weyl. Stereoselective Synthesis, D.4, Vol. 8, G. Helmechen, R. W. Hoffmann, J. Mulzer, E. Schaumann, ed.; Thieme, Stuttgart, 1996; p. 4747–4749 Search PubMed.
- (a) A similar method was employed in the synthesis of tetrodotoxin and its related compounds in this laboratory. For reading references, see: T. Nishikawa, M. Asai, N. Ohyabu, N. Yamamoto and M. Isobe, Angew. Chem., Int. Ed., 1999, 38, 3081–3084 CrossRef CAS; (b) M. Asai, T. Nishikawa, N. Ohyabu, N. Yamamoto and M. Isobe, Tetrahedron, 2001, 57, 4543–4558 CrossRef CAS; (c) T. Nishikawa, D. Urabe, K. Yoshida, T. Iwabuchi, M. Asai and M. Isobe, Org. Lett., 2002, 4, 2679–2682 CrossRef CAS; (d) T. Nishikawa, D. Urabe, K. Yoshida, T. Iwabuchi, M. Asai and M. Isobe, Chem.–Eur. J., 2004, 10, 452–462 CrossRef CAS.
- (a) H. Iio, M. Isobe, T. Kawai and T. Goto, Tetrahedron, 1979, 35, 941–948 CrossRef CAS; (b) Y.-S. Hon, S.-W. Lin, L. Lu and Y.-J. Chen, Tetrahedron, 1995, 51, 5019–5034 CrossRef CAS.
- A similar diastereoselectivity was observed in the reaction with MeMgBr in Et2O and MeLi in THF or Et2O.
- (a) J. D. Albright and L. Goldman, J. Am. Chem. Soc., 1965, 87, 4214–4216 CrossRef CAS; (b) J. D. Albright and L. Goldman, J. Am. Chem. Soc., 1967, 89, 2416–2423 CrossRef CAS.
- For deprotection of trichloroacetamide with NaBH4, see: F. Weygand and E. Frauendorfer, Chem. Ber., 1970, 103, 2437–2449 CrossRef CAS.
- (a) J.-L. Luche, J. Am. Chem. Soc., 1978, 100, 2226–2227 CrossRef CAS; (b) A. L. Gemal and J.-L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- The diastereomeric ratios of the reduction with NaBH4 in the presence of CeCl3, SmCl3 and YbCl3 were 13:10 = 7:1, 1.9:1 and 2.5:1, respectively.
- R. S. Tipson and A. Cohen, Carbohydr. Res., 1965, 1, 338–340 CrossRef CAS.
- When the formate 20 was treated with aq. Na2SO3, the formyl group was removed. However, the resulting α-hydroxy aldehyde 19 was found to be sensitive under the basic conditions of the next acetylide addition, probably because the retro-aldol reaction of the α-hydroxyl aldehyde 19 occurred.
- (a) For reviews of 1,3-dipolar cycloaddition between nitrones and acetylenes, see: J. P. Freeman, Chem. Rev., 1983, 83, 241–261 CrossRef CAS; (b) T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2010, 18, 3363–3376 CrossRef.
- J. E. Baldwin, R. G. Pudussery, A. K. Qureshi and B. Sklarz, J. Am. Chem. Soc., 1968, 90, 5325–5326 CrossRef CAS.
- We propose that the ring contraction (isomerization) of an isoxazoline to an aziridine aldehyde be called Baldwin rearrangement, because of the discovery by J. E. Baldwin in 1968.35 E. Vrancken, J.-M. Campagne and co-workers used the Baldwin rearrangement for this reaction in the following paper: E. Gayon, O. Debleds, M. Nicouleau, F. Lamaty, A. van der Lee, E. Vrancken and J.-M. Campagne, J. Org. Chem., 2010, 75, 6050–6053 CrossRef CAS.
- For an example of a similar spontaneous 1,3-dipolar cycloaddition between nitrone and acetylene followed by the Baldwin rearrangement, see: A. Padwa, M. Meske and Z. Ni, Tetrahedron Lett., 1993, 34, 5047–5050 CrossRef CAS.
- The 1,3-dipolar cycloaddition proceeded very slowly at room temperature. Under these conditions, isoxazoline 29 was not detected.
- The geometry of the nitrone moiety was determined to be Z by observing NOESY correlation between the protons at the C-3 position and the ortho position of the phenyl group.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21060f/ |
| This journal is © The Royal Society of Chemistry 2012 |