Electrostatic interaction based hollow Pt and Ru assemblies toward methanol oxidation†
Feng
Ye
a,
Jinhua
Yang
b,
Weiwei
Hu
a,
Hui
Liu
a,
Shijun
Liao
c,
Jianhuang
Zeng
*c and
Jun
Yang
*ab
aState Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: jyang@mail.ipe.ac.cn; Fax: 86-10-8254 4814; Tel: 86-10-8254 4915
bInstitute of Bioengineering and Nanotechnology, The Nanos, 31 Biopolis Way, Singapore 138669, Singapore
cSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangdong Key Laboratory for Fuel Cell Technology, Guangzhou 510641, China. E-mail: cejhzeng@scut.edu.cn; Fax: 86-20-3909 9665; Fax: 86-20-3909 9665
First published on 13th June 2012
Abstract
Mastery over the structure and/or composition of metal nanoparticles is an effective way to improve their catalytic activity on a mass basis. Herein, we report a facile solution route for the assembly of hollow Pt nanospheres (hPt) and ultrafine Ru nanoparticles based on electrostatic interactions. In this approach, negatively charged hollow Pt nanospheres with an average size of 12 nm and positively charged ultrafine Ru nanoparticles with an average size of 0.9 nm are first prepared, followed by the successful fabrication of hPt–Ru assemblies upon mixing the particles with opposite charges. The hPt–Ru assemblies at a Pt/Ru molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exhibit superior catalytic activity toward methanol oxidation in direct methanol fuel cells for the presence of a mixed-phase containing Pt and an effective oxophilic metal, and a smaller dilution effect on the Pt surface induced by Ru in the assemblies. This study offers a vivid example to demonstrate the integration of a second oxophilic metal into the active Pt catalyst capable of enhancing its catalytic properties by means of a physical construction.
:1 exhibit superior catalytic activity toward methanol oxidation in direct methanol fuel cells for the presence of a mixed-phase containing Pt and an effective oxophilic metal, and a smaller dilution effect on the Pt surface induced by Ru in the assemblies. This study offers a vivid example to demonstrate the integration of a second oxophilic metal into the active Pt catalyst capable of enhancing its catalytic properties by means of a physical construction.
1 Introduction
The strong growing interest in using direct methanol fuel cells (DMFCs) as portable and mobile power sources is rooted in their desirable features, such as a relatively small environmental footprint, compact system design, and higher volumetric energy densities compared with existing technologies.1,2 It has been well recognized that the success of DMFC technology depends largely on the electro-catalysts. Currently platinum-based nanomaterials are the most effective electro-catalysts to facilitate both anodic (methanol oxidation reaction, MOR) and cathodic (oxygen reduction reaction, ORR) reactions in DMFCs.3–6 However, the commercialization of DMFCs still faces a number of challenges at present: the high cost of the precious metal platinum (Pt) due to the scale of the Pt crystallites,7 low activities of the anode and cathode catalysts at room temperature,8–10 crossover of methanol from the anode to the cathode through the polymer electrolyte membrane (PEM),11–13 which interferes with oxygen reduction at the cathode, resulting in the creation of a mixed potential and a drastic decrease in DMFC performance.14 In order to overcome these barriers, it is necessary to maximize the activity of Pt-based catalysts by engineering their structure, morphology and/or composition.A number of approaches/techniques have been developed to resolve these critical issues for improving the performance of Pt-based electrocatalysts for the MOR and ORR in DMFCs. The most common practice is to prepare Pt-based electrocatalysts with a hollow interior or porous nanostructure,15–25 which have been demonstrated to have higher MOR and/or ORR activities than solid Pt particles. For instance, Liang et al. showed that Pt hollow nanospheres are twice as active as solid Pt nanoparticles of roughly the same size for methanol oxidation.16 The increase in activity could be attributed mainly to the larger surface area of the hollow structure, where the porous shell allows the internal surface of the catalyst to be accessible to the reactants. Although these nanostructures have displayed excellent catalytic properties, it is generally accepted that Pt alone has rather poor activity for methanol oxidation. At room and moderate temperatures, platinum could be readily poisoned by carbon monoxide (CO), an intermediate product of methanol oxidation.26–28 Without a second oxophilic metal, Pt itself would have to dissociate water to react away the CO intermediate at higher potentials. Herein, we report a facile, aqueous route to fabricate the assemblies of hollow Pt (hPt) and ultrafine Ru nanoparticles through electrostatic interaction, and their applications for catalyzing methanol oxidation, the anodic reaction in DMFCs. In this approach, negatively charged hollow Pt nanospheres with an average size of 12 nm and positively charged ultrafine Ru nanoparticles with an average size of 0.9 nm were first prepared. Subsequently, they were mixed together and assemblies of hPt and Ru were formed upon the electrostatic attraction between particles with opposite charges. Compared with the commercial PtRu alloy nanoparticles, the hPt–Ru assemblies have displayed superior catalytic activity toward methanol oxidation in DMFCs because of the presence of a mixed-phase containing Pt and an effective oxophilic metal. Oxygen-containing species can be preferentially formed on the Ru sites because Ru is more able to dissociate water at lower potentials, and the absorbed CO intermediate on Pt is then oxidized on the Ru sites after surface diffusion. This study offers a vivid example to demonstrate the integration of a second oxophilic metal into the active Pt catalyst capable of enhancing its catalytic properties by means of a physical construction.
2 Experimental
2.1 General materials
The chemical reagents, including silver nitrate (AgNO3, 99%), potassium platinum(II) chloride (K2PtCl4, 98%), sodium citrate dehydrate (Na3C6H5O7·2H2O, ≥99%), ruthenium(III) chloride trihydrate (RuCl3·3H2O, technical grade), sodium borohydride (NaBH4, 98%), and Nafion 117 solution (5% in a mixture of lower aliphatic alcohols and water) from Sigma-Aldrich, ethanol (ACS reagent, 99.5%) and methanol (ReagentPlus, 99%) from Merck, bis(p-sulfonatophenyl) phenylphosphane dihydrate dipotassium salt (BSPP, 97%) from Strem Chemicals, aqueous HClO4 solution (70%, ACS reagent) and Vulcan XC-72 carbon powders (BET surface area 250 m2 g−1 and average particle size 40–50 nm) from Cabot, were used as received. Deionized water was processed by a Milli-Q water purification system. All glassware and Teflon-coated magnetic stirring bars were cleaned with aqua regia, followed by copious rinsing with the Milli-Q water before drying in an oven.2.2 Synthesis of negatively charged Pt nanospheres (hPt)
The negatively charged hPt nanospheres were prepared using a published protocol with slight modification, in which Ag nanoparticles were used as sacrificial templates.17 Citrate-stabilized Ag nanoparticles with an average diameter of approximately 10 nm (seed particles) were firstly synthesized by the NaBH4 reduction of AgNO3.29 Briefly, an aqueous solution of AgNO3 (1 mM, 50 ml) was mixed with 2 ml of a 100 mM aqueous sodium citrate solution used as a stabilizer. Then 1 ml of a 100 mM aqueous NaBH4 solution was added dropwise under vigorous stirring, giving rise to a yellowish-brown hydrosol. The Ag hydrosol was aged for 24 h to decompose the residual NaBH4 before it was used in subsequent steps.For the synthesis of hPt nanospheres, the Ag hydrosol prepared above was refluxed at 110 °C for 10 min, and then 1 ml of a 50 mM aqueous K2PtCl4 solution, together with 2 ml of a 100 mM aqueous sodium citrate solution, was added. The mixture was refluxed for one more hour, and a reddish-brown solution of core–shell Ag–Pt nanoparticles was obtained. Subsequently, 30 mg of solid BSPP was added to the core–shell Ag–Pt hydrosol, and the mixture was aged for 4 h or more to completely remove the Ag core, resulting in the formation of hPt nanoparticles.
2.3 Synthesis of positively charged ultrafine Ru nanoparticles
The preparation of positively charged Ru nanoparticles proceeded according to a previously-reported approach.30 In a typical experiment, a 100 mM aqueous solution of NaBH4 was introduced dropwise into 50 ml of a 1 mM aqueous solution of RuCl3 under vigorous stirring. The volume of the NaBH4 solution was carefully controlled to maintain the pH value of the reaction system to be always lower than 4.9. The Ru hydrosol prepared in this way was very stable, as no precipitation ever took place, even after storage for 3 months. The Ru hydrosol was also aged for 24 h to decompose the residual NaBH4 for the following particle assembly.2.4 Preparation of hPt–Ru assemblies
For the assembly of hollow Pt nanospheres (hPt) and Ru nanoparticles based on electrostatic interaction at room temperature, the hPt hydrosol and Ru hydrosol as-prepared above were mixed together under vigorous stirring. The assemblies at different hPt/Ru molar ratios (2:1, 1:1, and 1:2, precursor ratio) were achieved by varying the volumes of the hPt and Ru hydrosols.
2.5 Characterizations of nanoparticles
Transmission electron microscopy (TEM) was performed on a FEI Tecnai G2 F20 electron microscope operated at 200 kV with the software package for automated electron tomography. The samples for TEM and high-resolution TEM (HRTEM) analysis were prepared by dispensing a drop of nanoparticle solution onto a 3 mm carbon-coated copper grid. Excess solution was removed by an absorbent paper, and the samples were dried in air at room temperature. The average particle size was obtained from a few randomly chosen areas in the TEM image containing approximately 100 nanoparticles each.An energy-dispersive X-ray spectroscopy (EDX) analyzer attached to the FEI Tecnai G2 F20 TEM operating in the scanning transmission electron microscopy (STEM) mode was used to analyze the components in the hPt–Ru assemblies. The electron beam was only 0.7 nm in diameter, capable of providing a high-resolution analysis.
Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/Max-3B diffractometer, using Cu-Kα radiation (λ = 1.54056 Å). X-ray photoelectron spectra (XPS) analyses were conducted on an ESCALAB MKII spectrometer (VG Scientific) using Al-Kα radiation (1486.71 eV). Samples for XRD and XPS were collected from hydrosols using centrifugation. They were washed with methanol several times and then dried at room temperature in a vacuum.
2.6 Electrochemical measurements
Electrochemical measurements were carried out in a standard three-electrode cell, which was connected to an AUTOLAB PGSTAT 30 potentiostat. A leak-free Ag/AgCl electrode (saturated with KCl) was used as the reference. The counter electrode was a platinum mesh (1 × 1 cm2) attached to a platinum wire.For loading the catalysts (hPt and hPt–Ru assemblies) on Vulcan XC-72 carbon supports, the calculated carbon powder was introduced into the hydrosol of the nanoparticles. After 24 h stirring, the nanoparticle/C (20 wt% Pt on carbon support) was collected using centrifugation and washed 3 times with methanol. They were then dried at room temperature under vacuum.
A thin layer of Nafion-impregnated catalyst cast on a vitreous carbon disk was used as the working electrode. The preparation of the catalyst ink followed a typical procedure in the literature with modification.7 10 mg of nanoparticle/C (hPt/C, or hPt–Ru/C) was ultrasonically dispersed in 10 ml aqueous solution containing 4 ml ethanol and 0.1 ml of the Nafion solution. A calculated volume of the ink was dispensed onto the 5 mm glassy carbon disk electrode to produce a nominal catalyst loading of 20 μg cm−2 (Pt base). The carbon electrode was then dried in a stream of warm air at 70 °C for 1 h.
The catalyst performance in the room temperature methanol oxidation reaction (MOR) was evaluated by cyclic voltammetry. The potential window from 0 V to 1 V was scanned at 20 mV s−1 until a stable response was obtained before the voltammograms were recorded. The electrolyte was 1 M methanol in 0.1 M perchloric acid. The current densities were also normalized in reference to the geometric area of the glassy carbon electrode.
3 Results and discussion
The preparation of hPt–Ru assemblies using electrostatic interaction is outlined in Fig. 1. In the first step, negatively charged hollow Pt nanospheres and positively charged ultrafine Ru nanoparticles were synthesized. Then, upon mixing of the two types of particles with opposite charges, hPt–Ru assemblies were obtained as the dominant product based on the electrostatic attraction between them.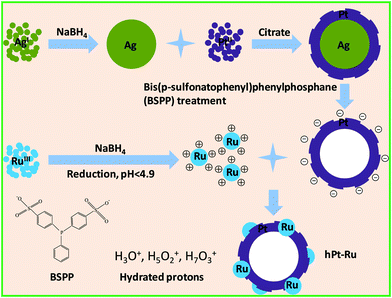 | ||
| Fig. 1 Schematic illustration for the assembly of hPt and Ru nanoparticles using electrostatic interaction. | ||
3.1 Negatively charged hollow Pt nanospheres and positively charged ultrafine Ru nanoparticles
We have discovered that Ag components could be reacted with BSPP to form a coordinating complex,17,31 which is central to the preparation of hPt nanospheres reported here. In this approach, core–shell Ag–Pt nanoparticles were first prepared by the successive reduction of Ag and Pt precursors, followed by the removal of Ag by BSPP, leaving behind hPt nanospheres with the same particle size as their core–shell predecessors. While concentrated HNO3 can also be used to remove Ag from the core–shell Ag–Pt nanoparticles, it usually induces aggregation of the nanoparticles as an undesirable side effect.The characterizations of hPt nanospheres have been well documented in our previous publication.17 The TEM and HRTEM images of the as-prepared hPt nanospheres with an average particle diameter of approximately 12 nm are shown in Fig. 2a and 2b. The hollow section in the core regions can be identified clearly by the contrast in brightness. The successful synthesis of the Pt nanophase could be demonstrated by XRD analysis. As seen in Fig. S1a of the ESI,† the lines of the {111} (2θ = 39.3°), {200} (2θ = 45.6°), {220} (2θ = 66.5°), and {311} (2θ = 80.1°) diffractions of the Pt were clearly identifiable, confirming that the hPt nanospheres have a face-centered cubic (fcc) lattice structure (JCPDS card No. 882343).32
 | ||
| Fig. 2 TEM (a,c) and HRTEM images (b,d) of the as-prepared negatively charged hollow Pt nanospheres (a,b) and positively charged ultrafine Ru nanoparticles (c,d). | ||
A comparison of TEM images of hPt (Fig. 2) and core–shell Ag–Pt nanoparticles (ESI, Fig. S2†) showed virtually no change in the particle size. The removal of the Ag component from the core–shell Ag–Pt nanoparticles therefore did not lead to the collapse of the spherical geometry. The interplanar spacing of approximately 0.23 nm indicated in the HRTEM image of the hPt nanospheres (Fig. 2b) corresponds to the {111} planes of fcc Pt. Additional structural details of hPt nanospheres can also be revealed by the microscopy images in Fig. 2. The discontinuous Pt shell with an average thickness of 1.6 nm, observed clearly after the removal of the Ag core, is of great importance, as it permits penetration of the reactants to access the inner surface of the hPt particles. It should be noted that BSPP served not only as the reactant to remove the Ag component but also as the substitute stabilizer for the initially citrate-stabilized core–shell particles. The sulfonic groups on BSPP imparted negative charges to the surface of the hPt particles, preventing them from aggregation through electrostatic repulsion.33
NaBH4 is a common reducing agent used to prepare metal nanoparticles from metal ions.34–37 It was also used in this work to prepare positively charged ultrafine Ru nanoparticles under carefully controlled experimental conditions.30 By means of the dropwise addition of 0.1 M aqueous NaBH4 solution to 10 ml of a 1 mM aqueous RuCl3 solution, a brown transparent hydrosol of ruthenium was obtained without any perceivable precipitation when the pH value of the reaction system was lower than 4.9. The reduction of ruthenium(III) by NaBH4 at room temperature may be categorically represented by the stoichiometric equation: 8Ru3+ + 3BH4− + 12H2O → 8Ru + 3B(OH)4− + 24H+.30,38 Although the aqueous NaBH4 solution is alkaline in character, the hydrogen ions generated in the reduction reaction can moderate the pH increase following the introduction of NaBH4. It has been speculated that the Ru hydrosol prepared by the above procedure was stabilized by surface adsorption of hydronium ions or other hydrated protons such as H5O2+ and H7O3+. The adsorption of hydronium ions and other hydrated protons on metal surfaces has been investigated both theoretically39 and experimentally.40,41 The hydronium ions and hydrated protons would impart a positive charge to the Ru nanoparticles and stabilize them against agglomeration by electrostatic repulsion. Fig. 2c and 2d show the TEM and HRTEM images of the as-prepared positively charged Ru nanoparticles. The average particle size of 0.88 nm was accompanied by a narrow size distribution with standard deviation 0.21 nm. Because of the tiny size, it was difficult to determine the interplanar spacing of the lattice planes from the HRTEM images of these ultrafine particles. The structural information could be revealed by the XRD pattern of the Ru particles (ESI, Fig. S1e†), which shows the broad “peaks” because of the ultrafine cluster size. The Bragg angles of 2θ at 38.6° and 44.0° are consistent with the {100} and {101} diffractions of the hexagonal Ru phase (JCPDS card No. 894903).32 The particle size calculated from the {101} line (2θ = 44.0°) using the Debye–Scherrer formula42,43 was about 0.92 nm, which is in good agreement with the TEM observation.
3.2 hPt–Ru assemblies based on the electrostatic interaction
Upon mixing of the hPt and Ru hydrosols at room temperature, hPt–Ru hetero-assemblies were found to be the dominant product because of the electrostatic attraction between sulfonic groups with negative charges and hydronium ions with positive charges. As indicated by Fig. S1b, c, and d (ESI† for the hPt–Ru assemblies recovered from the hydrosols, the XRD patterns displayed two distinct metal phases, which could be indexed to the fcc Pt and hexagonal Ru, respectively. In addition, a comparison of the XRD patterns of hPt–Ru assemblies with different Pt/Ru molar ratios (ESI, Fig. S1b, c, and d†) showed that the diffraction signals of the Ru phase were enhanced with the increase of the Ru molar ratio in the assemblies. In comparison with the commercial alloy PtRu/C catalysts, the XRD pattern of which showed a homogeneously mixed crystal lattice (ESI, Fig. S1f†), the presence of different crystal lattices demonstrated that the hPt–Ru assemblies were a physical mixture of hollow Pt nanospheres and ultrafine Ru nanoparticles. However, isolated hPt nanospheres or ultrafine Ru nanoparticles were not observed, indicating that the two types of particles with opposite charges on their surfaces have a strong tendency to bind together under experimental conditions. Fig. 3 shows the TEM and HRTEM images of the as-prepared hPt–Ru assemblies at different Pt/Ru molar ratios. A comparison of the TEM images of hPt–Ru assemblies (Fig. 3a, c, and e) and hPt nanoparticles (Fig. 2a) shows that the assembly with ultrafine Ru nanoparticles therefore did not lead to an apparent change in particle size of the hPt nanospheres. However, the comparison between the microscopy images of the hPt nanospheres (Fig. 2) and those of the hPt–Ru assemblies (Fig. 3) illustrates clearly that the assembly with ultrafine Ru nanoparticles using the electrostatic interaction resulted in a significant change in surface roughness of the hPt particles, and the degree of roughness increased with increasing molar ratio of Ru in the hPt–Ru assemblies.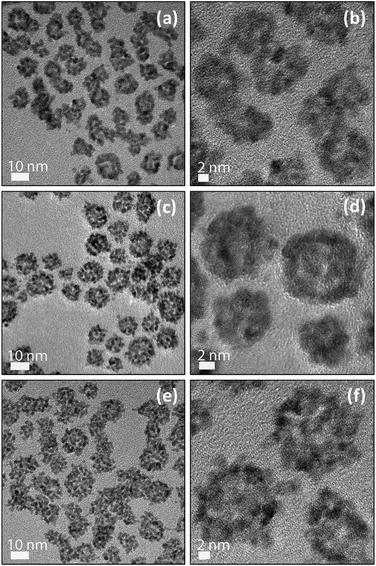 | ||
| Fig. 3 TEM (a,c,e) and HRTEM images (b,d,f) of the electrostatic interaction based hPt–Ru assemblies at Pt/Ru molar ratios of 2:1 (a,b), 1:1 (c,d), and 1:2 (e,f), respectively. | ||
To further confirm that the ultrafine Ru particles have been successfully assembled with hPt nanospheres in the mixture of hPt and Ru hydrosols, an EDX analyzer attached to the FEI Tecnai G2 F20 TEM operating in the high-angle scanning TEM (STEM) mode was used to analyze the components in an arbitrary single particle obtained from the mixture of hPt and Ru hydrosols. The electron beam was only 0.7 nm in diameter, capable of providing a high-resolution analysis. Fig. 4 shows the STEM image of hPt–Ru assemblies at a Pt/Ru molar ratio of 1:1, in which the coarse surface of the particles was also clearly observed. The inset is the EDX spectrum of the particle circled in the STEM image, which distinctly illustrates that the assembled particle is composed of Pt and Ru. Three sets of consistent data were obtained (ESI, Fig. S3†), demonstrating the reliability and reproducibility of the EDX analysis.
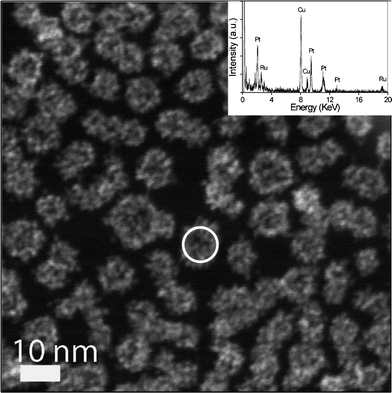 | ||
| Fig. 4 STEM image of electrostatic interaction based hPt–Ru assemblies at a Pt/Ru molar ratio of 1:1. Inset is the EDX analysis of an arbitrary assembled particle circled in the STEM image. | ||
3.3 Electrochemical activity of hPt–Ru assemblies
The hPt–Ru assemblies were examined for their electrocatalytic activity toward room-temperature methanol oxidation, and benchmarked against that in a commercial E-TEK PtRu/C catalyst (supported on XC-72, 20 wt% and Pt:Ru = 1:1). Voltammograms of methanol oxidation were obtained in the potential window 0–1 V at a swept rate of 20 mV s−1 (Fig. 5a). The current densities in the voltammograms were normalized in reference to the geometric area of the glassy carbon electrode. The voltammetric features were typical of methanol electrooxidation reported in the literature:44–46 methanol oxidation commenced at ∼0.3 V over the Pt catalysts, and a fully developed oxidation peak was formed at ∼0.7 V. The peak current densities associated with methanol oxidation in the forward and reverse scans were summarized in the ESI, Table S1.† Comparison between the current densities indicated that the hPt–Ru assemblies at a Pt/Ru molar ratio of 2:1 showed a higher catalytic activity than those of hPt–Ru assemblies at other ratios and commercial PtRu/C catalysts. The long-term performance of commercial PtRu/C catalysts and hPt–Ru assemblies in methanol oxidation is illustrated in the chronoamperograms in Fig. 5b. With the potential biased at 0.45 V, methanol was continuously oxidized on the catalyst surface and tenacious reaction intermediates such as CO would begin to accumulate if the kinetics of their removal could not keep pace with that of methanol oxidation.47 A more gradual decay of current density with time is therefore an indication of improved CO tolerance. As shown in Fig. 5b, the slower rate of decay for the hPt–Ru assemblies indicated their superior CO tolerance to that of the commercial PtRu/C catalysts and pure hPt nanospheres.
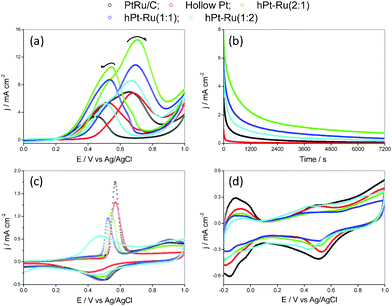 | ||
| Fig. 5 Cyclic voltammograms (a) of commercial PtRu/C, hPt, and hPt–Ru assemblies at different Pt/Ru ratios in argon-purged HClO4 (0.1 M) with methanol (1 M); chronoamperograms (b) of commercial PtRu/C, hPt, and hPt–Ru assemblies at different Pt/Ru ratios at 0.45 V vs. Ag/AgCl at room temperature in argon-purged HClO4 (0.1 M) with methanol (1 M); room-temperature CO stripping (c) from the commercial PtRu/C, hPt, and hPt–Ru assemblies at different Pt/Ru ratios in HClO4 (0.1 M); cyclic voltammograms (d) of commercial PtRu/C, hPt, and hPt–Ru assemblies at different Pt/Ru ratios in argon-purged HClO4 (0.1 M) at room temperature. | ||
Based on the bi-functional catalysis, the commonly accepted mechanism for methanol electrooxidation on Pt based catalysts,48–50 the presence of Ru, the most effective oxophilic metal, would facilitate the removal of CO from the Pt sites. The enhanced performance of Pt by Ru in methanol electrooxidation can be understood as a division of labor between the Pt and Ru sites: oxygen-containing species are preferentially formed on the Ru sites because Ru is more able to dissociate water at lower potentials. The adsorbed CO intermediate on Pt is oxidized at the pair-sites or on the Ru sites after surface diffusion. This is shown in Fig. 5c for the CO stripping voltammograms of commercial PtRu/C catalysts and hPt–Ru assemblies at different Pt/Ru molar ratios after the working electrode has been held at −0.15 V for 30 min in CO saturated 0.1 M HClO4. The CO stripping peaks of the hPt–Ru assemblies shifted to a more negative potential as compared to commercial PtRu/C catalysts and pure hPt nanospheres, indicating a more facile CO removal, and hence an improved CO tolerance in practice.
Although the CO stripping peaks of the hPt–Ru assemblies at Pt/Ru molar ratios of 1:1 and 1:2 were located at more negative potentials than that of hPt–Ru assemblies at a Pt/Ru molar ratio of 2:1, suggesting a more facile CO removal from the Pt surfaces in these hPt–Ru assemblies, the high ratio of Ru in the assemblies would reduce the Pt surface area available for occurrence of the methanol oxidation reaction. The electrochemically active surface area (ECSA) of Pt in pure hPt, hPt–Ru assemblies, and commercial PtRu/C catalysts was determined using cyclic voltammetry. There are three distinct potential regions in the voltammograms (Fig. 5d): the hydrogen adsorption/desorption region (−0.2–0.1 V vs. Ag/AgCl), the double layer region (0.1–0.4 V), and the surface oxide (OHads) formation/stripping region (>0.4 V).51 The specific ECSA, based on the unit weight of Pt and calculated by integrating the charge associated with the hydrogen adsorption/desorption potential region after double-layer correction, was summarized in the ESI, Table S2.† As indicated, the decrease in the ECSA of Pt with increasing Ru molar ratio in the hPt–Ru assemblies was clearly demonstrated. For the pure hPt, and for the hPt–Ru assemblies at a Pt/Ru molar ratio of 2:1, 1:1, and 1:2, the ECSA values were 29.7, 25.3, 22.0, and 14.4 m2 g−1, respectively. At an appropriate Pt/Ru molar ratio (2:1) in the hPt–Ru assemblies, the removal of the CO intermediate balances the ECSA of Pt available for methanol oxidation, thus offering optimum catalytic activity. At other Pt/Ru molar ratios (1:1 and 1:2), the ECSA of Pt for the methanol oxidation reaction is too low and far from the optimal balance, thus the hPt–Ru assemblies may suffer from insufficient area for the catalytic reaction, therefore are less active than hPt–Ru assemblies at optimal Pt/Ru molar ratios.
To interpret the higher catalytic activities of the hPt–Ru assemblies than those of commercial PtRu/C catalysts, whose ECSA was calculated to be 36.4 m2 g−1 based on the voltammogram in Fig. 5d, the surface chemistry of Pt and Ru in these catalysts was subjected to XPS analysis. As shown in Fig. 6a, the 4f spectra of Pt in hPt–Ru assemblies at a Pt/Ru molar ratio of 2:1 and in commercial PtRu/C catalysts are quite similar. The Pt 4f spectra can be deconvoluted into two pairs of doublets, in which the more intense doublet (at 70.6 and 73.9 eV, respectively) corresponded to Pt0. The second and weaker doublet, with binding energies of approximately 1.4 eV higher than those of Pt0, could be assigned to PtII as in PtO and Pt(OH)2.52,53 On the other hand, the chemical states of Ru in hPt–Ru assemblies and commercial PtRu/C catalysts were significantly different. As shown in Fig. 6b, the 3p spectra of Ru in PtRu/C catalysts can also be deconvoluted into two pairs of doublets. The more intense doublet, at 461.0 and 483.2 eV, corresponded to the zerovalent state of Ru, while the weaker doublet, at 462.8 and 484.7 eV, could be assigned to RuIV (e.g. RuO2).46,52–54 However, for Ru in hPt–Ru assemblies, the 3p spectra were laden with noise and poorly resolved, making it difficult to quantify the chemical states of Ru. Nevertheless, the spectra can also be deconvoluted into two pairs of doublets, and the signal corresponding to Ru0 was much weaker than that of Ru in the oxidized state (Fig. 6b), suggesting that most of the Ru in the hPt–Ru assemblies was present as Ru oxides with binding energies of 462.8 and 484.7 eV. The presence of Ru oxides might be attributed to the tiny size of the Ru nanoparticles, which activated the particles to be oxidized at room temperature.
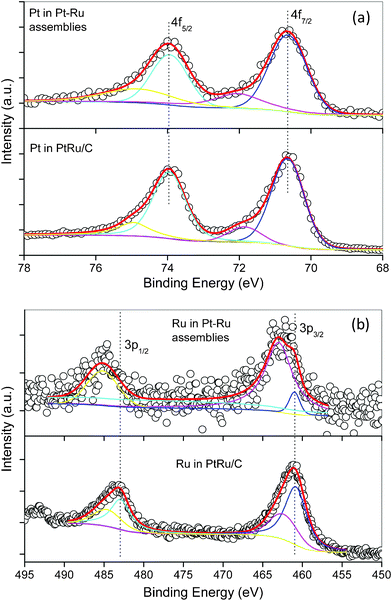 | ||
| Fig. 6 4f XPS spectra of Pt (a) and 3p XPS spectra of Ru (b) in hPt–Ru assemblies and commercial PtRu/C catalysts. | ||
The existence of Ru oxides in hPt–Ru assemblies would be favorable for methanol oxidation. The improved catalytic activity did not come from the smaller Pt particle size and higher ECSA, since hPt–Ru assemblies did not have either. However, the superior catalytic activity of the hPt–Ru assemblies for methanol oxidation was not surprising based on the reports by Rolison and co-workers,55,56 which discussed mixing hydrous ruthenium oxides with Pt to enhance the electro-catalytic activity for methanol oxidation. The homogeneous PtRu alloy catalysts were found to be orders of magnitude less active than mixed-phase electrocatalysts containing Pt metal and hydrous ruthenium oxides. The high activity of mixed-phase electrocatalysts was derived from the presence of hydrous ruthenium oxides, the electron and proton conducting materials, which were intimately mixed with Pt. Their experimental observations were subsequently confirmed by many other researchers.57–63 For example, Qiu and co-workers deposited Pt on preformed hydrous Ru oxide and observed enhanced activity for catalysis of the methanol oxidation reaction.60 The comparison between the alloy PtRu catalysts prepared from co-reduction of the metal precursors and from the mixing of preformed metal colloids was carried out by Lamy and co-workers.64 They concluded that alloyed catalysts had the lowest activity because of the migration of Ru to the alloy surface under the operating conditions, thereby reducing the Pt surface area available for methanol adsorption.
The smaller surface dilution effect may also account for the enhanced catalytic activity of hPt–Ru assemblies toward methanol oxidation. It has been generally accepted that methanol oxidation commences by methanol adsorption on multiple Pt sites (3-fold methanol adsorption sites, denoted as Pt3).11 The surface conditions of commercial PtRu/C catalysts and hPt–Ru assemblies can be schematically described by Fig. 7a and 7b, respectively. For the alloy PtRu/C catalysts, the atomic mixing and uniform distribution of Pt and Ru on the surface may inhibit the adsorption of methanol along the directions labeled 1 and 3 (inset of Fig. 7a), and therefore lead to negative effects on the catalytic activity for methanol oxidation. However, for the hPt–Ru assemblies, several closely neighboring Pt atoms share an ultrafine Ru cluster on the surface, not only making use of the oxygen-containing species brought by the Ru cluster, but also allowing the adsorption of methanol in all directions (inset of Fig. 7b). Compared with the surface dilution of Pt induced by alloying with Ru, the hPt–Ru assemblies can maintain good catalytic activity toward methanol oxidation at suitable Pt/Ru molar ratios (for example, Pt/Ru of 2:1 in this work), although the coherent interfaces between the hPt and Ru nanoparticles in the assemblies resulted in some blockage of the surface area of the Pt domains.
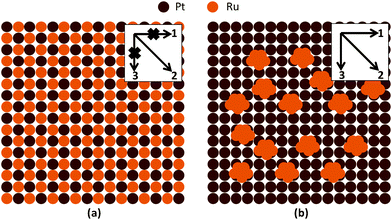 | ||
| Fig. 7 Schematic illustration of the surface conditions of alloy PtRu catalysts (a) and hPt–Ru assemblies (b). Insets show the possible direction for methanol adsorption on the surface of the catalysts. | ||
4 Conclusions
In summary, a facile solution route for the fabrication of hPt–Ru assemblies based on electrostatic interaction was reported. In this contribution, negatively charged hollow Pt nanospheres with an average size of 12 nm and positively charged ultrafine Ru nanoparticles with an average size of 0.9 nm were first prepared. Then the hPt–Ru assemblies were fabricated successfully upon mixing of the particles with opposite charges. The hPt–Ru assemblies have displayed superior catalytic activity toward methanol oxidation in DMFCs, which was attributed to the presence of Ru oxide because of its high electron and proton conductivity. The smaller surface dilution of Pt by ultrafine Ru clusters in hPt–Ru assemblies was also supposed to account for the enhanced catalytic activity of the assemblies toward methanol oxidation. In hPt–Ru assemblies, the closely neighboring Pt atoms share a ultrafine Ru cluster on the surface, not only making use of the oxygen-containing species brought by the Ru cluster, but also allowing sufficient adsorption of methanol on the surface. This study offers a vivid example to demonstrate the integration of a second oxophilic metal into the active Pt catalyst capable of enhancing its catalytic properties by means of a physical construction. By optimizing the chemical composition of the hPt–Ru assemblies through varying the ratio of Pt to Ru during the mixing process, further enhancement in activity for the methanol oxidation reaction could be expected.Acknowledgements
We are grateful to Prof. Jim Yang Lee (National University of Singapore) for the use of the XPS facilities. This work was supported by Institute of Bioengineering and Nanotechnlogy (Biomedical Research Council, Agency for Science, Technology and Research, Singapore), the 100 Talents Program of the Chinese Academy of Sciences, National Natural Science Foundation of China (Project No.: 21173226, 21106151), and Foundation of Guangdong Key Laboratory for Fuel Cell Technology.References
- E. Antolini, Mater. Chem. Phys., 2003, 78, 563 CrossRef CAS.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS.
- M. L. Perry and T. F. Fuller, J. Electrochem. Soc., 2002, 149, S59 CrossRef CAS.
- J. Chen, B. Lim, E. P. Lee and Y. Xia, Nano Today, 2009, 4, 81 CrossRef CAS.
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527 CrossRef CAS.
- N. M. Marković and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117 CrossRef.
- W. Chen, J. Kim, S. Sun and S. Chen, J. Phys. Chem. C, 2008, 112, 3891 CAS.
- H. A. Gasteiger, N. Marković, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1993, 97, 12020 CrossRef CAS.
- G. Vijayaraghavan, L. Gao and C. Korzeniewski, Langmuir, 2003, 19, 2333 CrossRef CAS.
- J. M. Wang, S. R. Brankovic, Y. Zhu, J. C. Hanson and R. R. Adzic, J. Electrochem. Soc., 2003, 150, A1108 CrossRef CAS.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, J. Electroanal. Chem., 2005, 580, 145 CrossRef CAS.
- Y. Ding, M. Chen and J. Erlebacher, J. Am. Chem. Soc., 2004, 126, 6876 CrossRef CAS.
- H. Liang, H. Zhang, J. Hu, Y. Guo, L. Wan and C. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540 CrossRef CAS.
- J. Yang, J. Y. Lee, H.-P. Too and S. Valiyaveettil, J. Phys. Chem. B, 2006, 110, 125 CrossRef CAS.
- X. Teng, X. Liang, S. Maksimuk and H. Yang, Small, 2006, 2, 249 CrossRef CAS.
- G. Chen, D. Xia, Z. Nie, Z. Wang, L. Wang, L. Zhang and J. Zhang, Chem. Mater., 2007, 19, 1840 CrossRef CAS.
- X. Teng, S. Maksimuk, S. Frommer and H. Yang, Chem. Mater., 2007, 19, 36 CrossRef CAS.
- H. M. Chen, R.-S. Liu, M.-Y. Lo, S.-C. Chang, L.-D. Tsai, Y.-M. Peng and J.-F. Lee, J. Phys. Chem. C, 2008, 112, 7522 CAS.
- S. Guo, S. Dong and E. Wang, Chem.–Eur. J., 2008, 14, 4689 CrossRef CAS.
- C. Xu, L. Wang, R. Wang, K. Wang, Y. Zhang, F. Tian and Y. Ding, Adv. Mater., 2009, 21, 2165 CrossRef CAS.
- J. Yang, E. H. Sargent, S. O. Kelley and J. Y. Ying, Nat. Mater., 2009, 8, 683 CrossRef CAS.
- Z. Peng, J. Wu and H. Yang, Chem. Mater., 2010, 22, 1098 CrossRef CAS.
- V. S. Bagotzky, Y. B. Vassiliev and O. A. Khazova, J. Electroanal. Chem., 1977, 81, 229 CrossRef.
- J. B. Goodenough, A. Hamnett, B. J. Kennedy, R. Manoharan and S. A. Weeks, Electrochim. Acta, 1990, 35, 199 CrossRef CAS.
- R. Parsons and T. VanderNoot, J. Electroanal. Chem., 1988, 257, 9 CrossRef CAS.
- J. Zeng, J. Yang, J. Y. Lee and W. Zhou, J. Phys. Chem. B, 2006, 110, 24606 CrossRef CAS.
- J. Yang, J. Y. Lee, T. C. Deivaraj and H.-P. Too, J. Colloid Interface Sci., 2004, 271, 308 CrossRef CAS.
- Y.-N. Tan, J. Yang, J. Y. Lee and D. I. C. Wang, J. Phys. Chem. C, 2007, 111, 14084 CAS.
- W. F. McClune, Powder Diffraction File Alphabetical Index Inorganic Phase, JCPDS, Swarthmore, PA, 1980 Search PubMed.
- J. Zhong, J. Qu, F. Ye, C. Wang, L. Meng and J. Yang, J. Colloid Interface Sci., 2011, 361, 59 CrossRef CAS.
- G. N. Glavee, K. J. Klabunde, C. M. Sorensen and G. C. Hadjapanayis, Langmuir, 1992, 8, 771 CrossRef CAS.
- G. N. Glavee, K. J. Klabunde, C. M. Sorensen and G. C. Hadjapanayis, Langmuir, 1993, 9, 162 CrossRef CAS.
- J. Yang, J. Y. Lee, T. C. Deivaraj and H. P. Too, J. Colloid Interface Sci., 2004, 277, 95 CrossRef CAS.
- J. Yang, J. Y. Lee, H.-P. Too, G.-M. Chow and L. M. Gan, Chem. Phys., 2006, 323, 304 CrossRef CAS.
- S. Chen and K. Kimura, Langmuir, 1999, 15, 1075 CrossRef CAS.
- P. P. Olivera, A. Ferral and E. M. Patrito, J. Phys. Chem. B, 2001, 105, 7227 CrossRef CAS.
- K.-I. Ataka, T. Yotsuyanagi and M. Osawa, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- N. Chen, P. Blowers and R. I. Masel, Surf. Sci., 1999, 419, 150 CrossRef CAS.
- V. Radmilovic, H. A. Gasteiger and P. N. Ross, J. Catal., 1995, 154, 98 CrossRef CAS.
- K.-W. Park, J.-H. Choi, B.-K. Kwon, S.-A. Lee and Y.-E. Sung, J. Phys. Chem. B, 2002, 106, 1869 CrossRef CAS.
- M. L. Anderson, R. M. Stroud and D. R. Rolison, Nano Lett., 2002, 2, 235 CrossRef CAS.
- J. F. Drillet, A. Ee, J. Friedemann, R. Kötz, B. Schnyder and V. M. Schmidt, Electrochim. Acta, 2002, 47, 1983 CrossRef CAS.
- X. Zhang and K.-Y. Chan, Chem. Mater., 2003, 15, 451 CrossRef CAS.
- J. Zeng and J. Y. Lee, J. Power Sources, 2004, 140, 268 CrossRef.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 275 CrossRef CAS.
- B. Gurau, R. Viswanathan, R. Liu, T. J. Lafrenz, K. L. Ley, E. S. Smotkin, E. Reddington, A. Sapienza, B. C. Chan, T. E. Mallouk and S. Sarangapani, J. Phys. Chem. B, 1998, 102, 9997 CrossRef CAS.
- V. Stamenkovic, T. J. Schmidt, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2002, 106, 11970 CrossRef CAS.
- Z. Liu, J. Y. Lee, M. Han, W. Chen and L. M. Gan, J. Mater. Chem., 2002, 12, 2453 RSC.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble, NIST Standard Reference Database 20, Version 3.2 (Web Version) Search PubMed.
- A. S. Aricò, P. Cretì, H. Kim, R. Mantegna, N. Giordano and V. Antonucci, J. Electrochem. Soc., 1996, 143, 3950 CrossRef.
- D. R. Rolison, P. L. Hagans, K. E. Swider and J. W. Long, Langmuir, 1999, 15, 774 CrossRef CAS.
- J. W. Long, R. M. Stroud, K. E. Swider and D. B. Rolison, J. Phys. Chem. B, 2000, 104, 9772 CrossRef CAS.
- H. M. Villullas, F. I. Mattos-Costa and L. O. S. Bulhőes, J. Phys. Chem. B, 2004, 108, 12898 CrossRef CAS.
- Z. Chen, X. Qiu, B. Lu, S. Zhang, W. Zhu and L. Chen, Electrochem. Commun., 2005, 7, 593 CrossRef CAS.
- F. Scheiba, M. Scholz, L. Cao, R. Schafranek, C. Roth, C. Cremers, X. Qiu, U. Stimming and H. Fuess, Fuel Cells, 2006, 6, 439 CrossRef CAS.
- L. Cao, F. Scheiba, C. Roth, F. Schweiger, C. Cremers, U. Stimming, H. Fuess, L. Chen, W. Zhu and X. Qiu, Angew. Chem., Int. Ed., 2006, 45, 5315 CrossRef CAS.
- L. P. R. Profeti, F. C. Simőes, P. Olivi, K. B. Kokoh, C. Coutanceau, J.-M. Léger and C. Lamy, J. Power Sources, 2006, 158, 1195 CrossRef CAS.
- S. Y. Huang, C. M. Chang, K. W. Wang and C. T. Yeh, ChemPhysChem, 2007, 8, 1774 CrossRef CAS.
- F. Peng, C. Zhou, H. Wang, H. Yu, J. Liang and J. Yang, Catal. Commun., 2009, 10, 533 CrossRef CAS.
- L. Dubau, F. Hahn, C. Coutanceau, J.-M. Léger and C. Lamy, J. Electroanal. Chem., 2003, 554–555, 407 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: XRD patterns of hPt nanospheres, hPt–Ru assemblies at different Pt/Ru molar ratios, Ru nanoparticles, and PtRu/C catalysts (Fig. S1), TEM and HRTEM images of core–shell Ag–Pt nanoparticles (Fig. S2), STEM image and EDX analyses of hPt–Ru assemblies at a Pt/Ru molar ratio of 1:1 (Fig. S3), and results of electrochemical measurements (Table S1 and Table S2). See DOI: 10.1039/c2ra21140h/ |
| This journal is © The Royal Society of Chemistry 2012 |