Electrochemical synthesis of a novel compound, 5-acetyl-2,9-decanedione, and theoretical analysis of its lithium ion complex
Israel
Cabasso
,
Mingyu
Li
and
Youxin
Yuan
*
The Michael Szwarc Polymer Research Institute, Chemistry Department, State University of New York-esf, Syracuse, NY 13210. E-mail: yxyuan@syr.edu
First published on 24th August 2012
Abstract
Reported is the electrosynthesis of a novel liquid compound, 5-acetyl-2,9-decanedione (TK) [n35d = 1.4524 and Tm < −70 °C, Tb > 200 °C], the molecule has unique hierarchic “Y” structure with three carbonyl groups anchored at the end of each arm. The TK is formed though the electrochemical reformation of bio-derived levulinic acid in MeOH/H2O system. The possible mechanism of formation of the TK via a side reaction of Kolbe electrolysis has been proposed. Theoretical analysis of the TK coordination with lithium ions, forming bidentate Li+ (TK)Li-O1O2, or Li+ (TK)Li-O2O3, or tridentate Li+ (TK)Li-O1O2O3 complexes has been conducted. The results suggest that one TK can possess two Li+ ions, thus, it might be a competitive solvent for lithium ion batteries.
Introduction
The preliminary computations done at this and other1–5 laboratories on common liquid solvents with a wide working temperature range in lithium ion battery (such as, propylene carbonate PC, and ethylene carbonate EC), found that these materials have a very high interaction ability with lithium ions while retaining their physical characteristic. The search for alternative less expensive solvents with the same or better properties has been a subject of interest for a long time.6–8 It is apparent that the synthetic approach should be linked to the production of a non-conversional molecule that accommodates the physical and chemical properties of such solvent, i.e., a wide working temperature range and multi-functional binding sites for lithium ion. We wish to report here the synthesis of a novel compound, 5-acetyl-2,9-decanedione (TK), by electrochemical reformation of levulinic acid, LA (known to be produced from biomass waste9–13) in MeOH/H2O system. To the best of our knowledge the synthesis of this compound has never been reported.The 5-acetyl-2,9-decanedione being liquid, with low melting point and high boiling temperature, has a unique hierarchic “Y” structure with three carbonyl groups anchored at the end of each arm. The preliminary results of theoretical computation of solvation of the 5-acetyl-2,9-decanedione with lithium ion provide insight into the electronic structure and thermodynamic properties such as the standards enthalpy, entropy and Gibbs energy change, and suggest that TK could be a potential solvent in lithium-ion battery technology.
We use electrochemical synthesis to produce such an unconventional compound as TK because the synthesis method is a one-pot reaction that is inherently “clean”, and consists of an unlimited supply of electrons (via electricity) as a “reagent”.14,15 As such, the old Kolbe electroorganic reaction16 which is highly efficient for decarboxylating alkanoic acids due to its specificity and multilateral flexibility. The reaction is initiated by electro-oxidation of the carboxylate anion, that decomposes into an alkyl radical (i.e., one-electron oxidation), and CO2. Consequently, two radicals combine to form a dimer, or by losing another electron form a carbenium ion (i.e., two-electron oxidation). The relative ratio of radicals and carbenium species depends on the electrode materials, the polarity of the solvent, the chemical structure of carboxylate acids and other electrolytic parameters.17–19 Irrespective of the mechanism both intermediates yield longer carbon chain products through an alkyl radical coupling, or via esterification of the carbenium ions with carboxylate anions.
Experimental
Materials
Levulinic acid, hexanoic acid, valeric acid and butyric acid (ACS grade Aldrich), anhydrous methyl alcohol (MeOH) and dichloromethane (CH2Cl2) (Mallinckrodt Chemicals), sodium metal (Na lump, in kerosene, 99% Aldrich), anhydrous sodium sulfate (Na2SO4) (EMD Chemicals Inc.), hexane and ethyl acetate (ACS grade), were used as received.General electrolysis procedures
The overall experimental procedure is illustrated in Scheme 1. A typical electrolysis of levulinic acid experiment consists of dissolving levulinic acid (25 mmol) and Na metal (0.5 mmol) in 50 mL mixture of methanol/water (0–100 mol%, at pH∼5). The reactant solution was placed in a water-jacketed electrolysis cell (100 mL) with two 12 × 15 × 0.05 mm pieces of platinum foils (Aldrich, 99.99%) serving as the anode/cathode pair and mounted in parallel with a gap of 1–2 mm. Electrolysis was achieved by applying either a constant dc current ∼0.45–0.7 A (current density ∼0.25–0.4 A cm−2) or a constant voltage 60–100 V. The solution temperature was maintained at 30 ± 1 °C. Electrolysis was terminated after the consumption of 1.2–2.0 F/mol of charge yielding a pH of ∼7. The resulting products were extracted with CH2Cl2 (3 × 20 ml) and dried over anhydrous Na2SO4.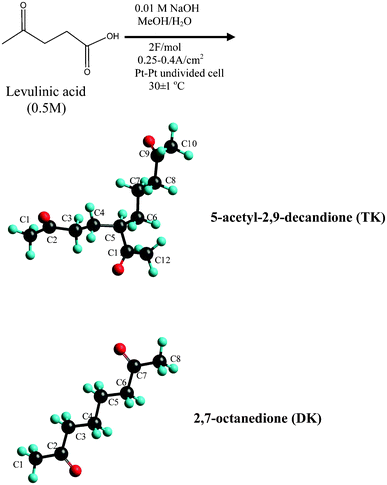 | ||
| Scheme 1 Electrochemical reformation of levulinic acid. | ||
The entire product mixture was separated using silica gel (Silica Gel 60, EM Science) column and hexane/ethyl acetate (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:2 by volume) mixture as the eluent and analyzed by GC-MS. The yield was determined by GC/FID using naphthalene as the internal standard. Each product was identified using high-resolution mass spectrometry (HRMS, ESI-TOF) and NMR spectroscopy.
:1 to 1:2 by volume) mixture as the eluent and analyzed by GC-MS. The yield was determined by GC/FID using naphthalene as the internal standard. Each product was identified using high-resolution mass spectrometry (HRMS, ESI-TOF) and NMR spectroscopy.
The geometries of all the stationary points including the Li+ complexes, Li+ and the ligands were optimized at restricted Hartree–Fock (RHF) level of theory with 6-31G basis set. All the stationary points were characterized by their positive harmonic vibrational frequencies as minima. The entropies, the enthalpies and the Gibbs free energies were calculated at RHF/6-31G as well. All the ab initio molecular orbital computation were performed with the GAUSSIAN 09 program package.20
Results and discussion
1) Synthesis and characterization of 5-acetyl-2,9-decanedione (TK)
The electrochemical reformation of LA in anhydrous MeOH produced ∼70 wt% of 2,7-octanedione. Apparently, this reaction follows an electro-oxidative decarboxylation radical coupling pathway. The presence of water gave rise to a second prominent novel product which appears as a yellowish liquid with a refractive index of n35d = 1.4524 (higher than 2,7-octanedione n35d = 1.4319) and a freezing point Tm < −70 °C, a boiling temperature Tb > 200 °C. The FTIR spectrum shows a strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1712 cm−1, υas CH3 at 2998 cm−1, υas CH2 at 2943 cm−1, δas CH3 at 1417 cm−1, δs CH3 1355 cm−1 and C–C(O)–C stretching and bending at 1227 cm−1.
O stretch at 1712 cm−1, υas CH3 at 2998 cm−1, υas CH2 at 2943 cm−1, δas CH3 at 1417 cm−1, δs CH3 1355 cm−1 and C–C(O)–C stretching and bending at 1227 cm−1.
The high-resolution mass spectrum found that the molecular mass 212.1410 Dalton is in agreement with the theoretical value of 212.1412 Dalton for the molecular formula of C12H20O3.
Major MS fragments (m/z) (Fig. 1): 194[M–H2O]+; 169[M–CH3CO]+; 141[CH3COCH2CH2CH2+CHCOCH3]; 127[CH3COCH2CH2+CHCOCH3]; 85[CH3COCH2CH2CH2+]; 71 [CH3COCH2CH2+]; 43[CH3CO+]. One and two dimensional 1H and 13C NMR experiments were used to deduce the structure of the proposed compound 5-acetyl-2,9-decanedione as follows: the 1D 1H NMR spectrum (Fig. 2) shows one methine, five methylenes, and three methyls. There are three methyl singlets that appear in the 2.08–2.11 ppm region which indicate three acetyl groups, other patterns were not first order and therefore analyzed by 2D COSY data.
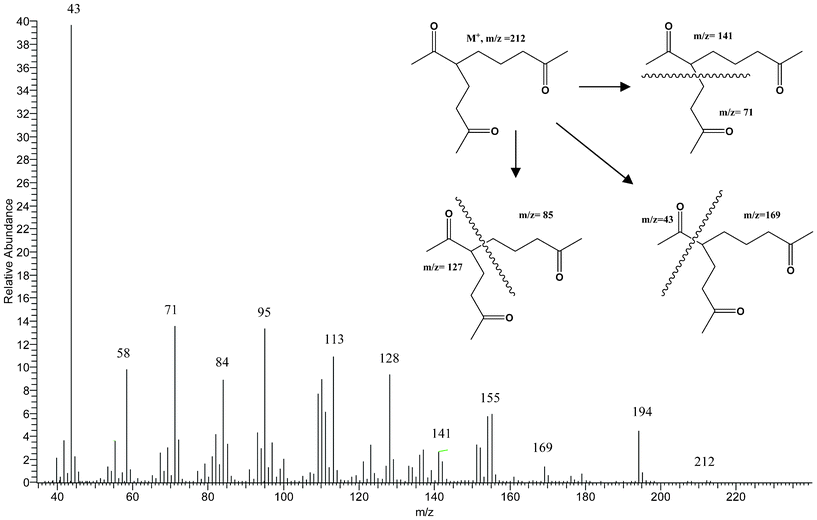 | ||
| Fig. 1 Mass spectrum of 5-acetyl-2,9-decanedione. | ||
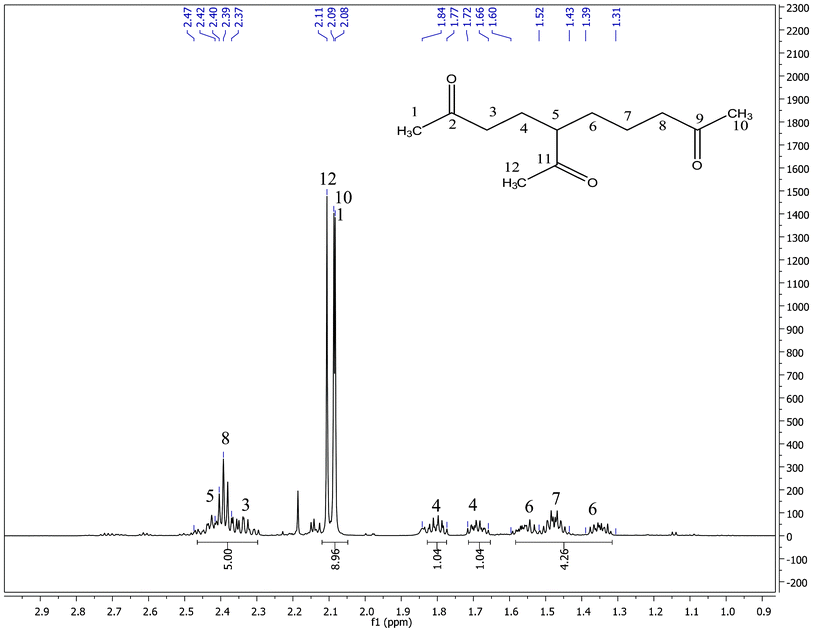 | ||
| Fig. 2 1H NMR spectrum of 5-acetyl-2,9-decanedione in CDCl3. | ||
13C NMR, δ (ppm) consists of three keto-carbons 211.71 (C11), 208.21(C9) and 207.89(C2); nine aliphatic carbons which can be attributed to one methine at 51.80 (C5), five methylene at 43.23(C8), 40.66 (C3), 30.52 (C6), 24.55 (C4), 21.23 (C7), and three methyls at 29.87 (C1), 29.91 (C10), 28.66 (C12) (Table 1). A detailed structure determination of 5-acetyl-2,9-decanedione was carried out by using 2D NMR spectroscopy (Fig. 3). The 1H–13C HSQC-DEPT edited NMR technique correlates the direct 1H–13C spin couplings in the molecule, as well as giving information on how many hydrogens are attached to a given carbon. This technique is very useful as the proposed structure has a chiral center at position C5 (δ 51.80 ppm in the 13C-NMR) that renders the neighboring methylene protons H-4 and H-6 diastereotopic, splitting the corresponding 1H-NMR signals into two paired multiplets; one pair at δ 1.66–1.73 ppm and δ 1.77–1.85 ppm (m, 2H, H-4) and the other pair at δ 1.31–1.39 ppm and δ 1.52–1.60 ppm (m, 2H, H-6). The 1H–1H COSY NMR confirms the H-5 and H-4, H-5 and H-6 couplings for the assumed structure. The linear six-carbon network C3 to C8 in the assumed structure were evidenced by couplings of the H-3 to H-4, H-4 to H-5, H-5 to H-6, H-6 to H-7, and H-7 to H-8 in the 1H-1H COSY spectra. These assignments were used to assign the 13C signals with the 2D-HSQC-DEPT data. Table 1 shows all assignments that were made from the 2D data. The remaining structural assignments are the three acetyl groups, the HMBC data confirmed the placement of these groups as follows: the CH3-1 correlates with the keto C2 and methylene C3; CH3-12 correlates with keto C11 and methine C5; and the CH3-10 correlates with the keto C9 and methylene C8. These three methyl group correlations support the connection to the carbonyl group as drawn in Scheme 1. Other long range HMBC correlations are listed in Table 1 which further supports the proposed structure.
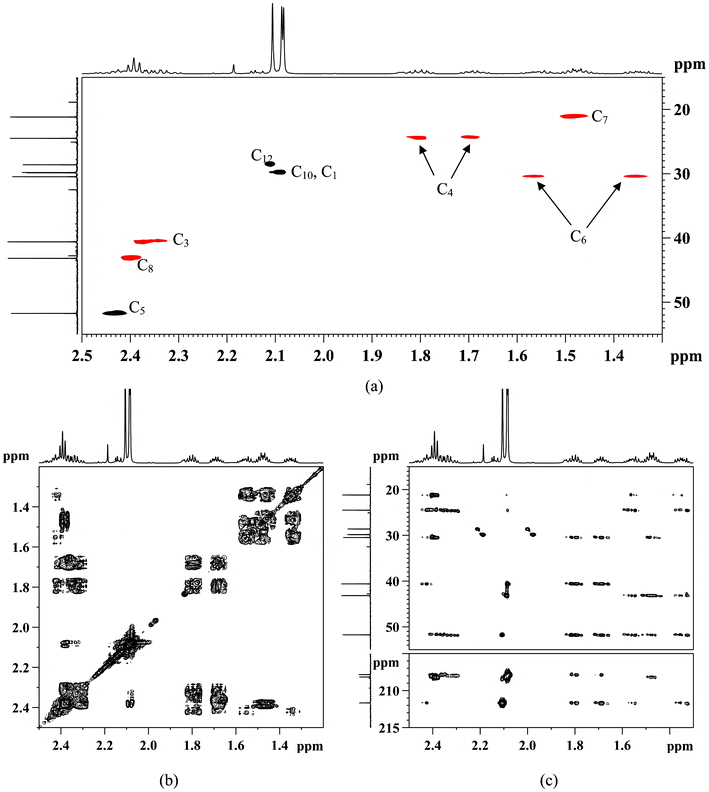 | ||
| Fig. 3 Characterization of 5-acetyl-2,9-decanedione with 2D-NMR in CDCl3: (a) 1H–13C HSQC-DEPT (black dots –CH and CH3; red dots –CH2); (b) 1H–1H COSY and (c) 1H–13C HMBC. | ||
 | ||
| Scheme 2 Proposed synthetic pathways toward 5-acetyl-2,9,-decanedione. | ||
| No. | δ C | δ H (mult.) | HMBC |
|---|---|---|---|
| 1 | 29.87 | 2.08(s) | C-2, 3 |
| 2 | 207.89 | ||
| 3 | 40.66 | 2.29–2.40(m) | C-2, 4, 5 |
| 4a | 24.55 | 1.66–1.73(m) | C-2, 3 ,5, 6, 11 |
| 4b | 1.77–1.85(m) | ||
| 5 | 51.80 | 2.39–2.47(m) | C-3, 4, 6, 7, 11 |
| 6a | 30.52 | 1.31–1.39(m) | C-4, 5, 7, 8, 11 |
| 6b | 1.52–1.60(m) | ||
| 7 | 21.23 | 1.43–1.52(m) | C-5, 6, 8, 9 |
| 8 | 43.23 | 2.37–2.42(m) | C-6, 7, 9 |
| 9 | 208.21 | ||
| 10 | 29.91 | 2.09(s) | C-8, 9 |
| 11 | 211.71 | ||
| 12 | 28.66 | 2.11(s) | C-5, 11 |
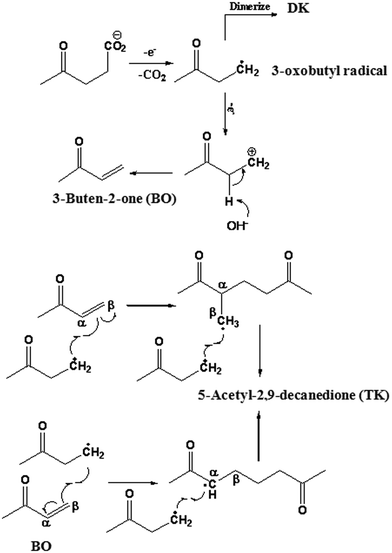
Scheme 2 illustrates the possible reaction pathway that leads to the formation of 5-acetyl-2,9-decanedione. The electrochemical reformation of levulinic acids starts with an initial discharge of levulinate ions at the anode, followed by decarboxylation forming 3-oxobutyl radicals. Subsequently, the recombination of the resulting radicals forms the Kolbe dimer (DK). However, 3-oxobutyl radicals can further undergo electro-oxidation at the anode, losing another electron to form the corresponding 3-oxobutyl carbenium ions. This reaction is favored in an aqueous medium due to higher ionization power21,22 and Brønsted base effect of H2O molecules. With the assistance of water molecules, a transient olefin product 3-buten-2-one (BO) would be formed through E1 elimination of a proton from the initial carbenium ion. Although other carbenium-derived products such as alcohol and ester are also possible, none of them were detected in this study. Most likely this is due to the electron-withdrawing induction effect of the keto group on the 3-oxobutyl cation. This induction increases the acidity of α-H and hence facilitates the E1 elimination products. The intermediate (BO) continuously reacts with the 3-oxobutyl radicals at the double bond from either the α or β position to form 5-acetyl-2,9-decanedione. It should be noted that the intermediate (BO) in the proposed synthetic pathways could not be isolated in the actual reaction mixture, apparently due to the fast reaction in radical addition step.
Thus, in order to verify the possibility of such mechanism, a commercially available product 3-buten-2-one was added during the electrochemical reformation of LA (with anhydrous MeOH as the solvent) and product distributions is illustrated in Scheme 3A. Aside from homo-coupling 2,7-octanedione (85.3% relative yield), the TK was found in a considerable yield (∼14.7%), as predicted by the proposed mechanism (Scheme 2). In fact, this reaction mechanism appears to be rather general and represents a novel approach for the synthesis of unique hierarchy “Y” structures (e.g., TK, Y-mer 1, and Y-mer 2) as we demonstrated by the electrochemical reformation of valeric acid (VA) in the presence of 3-buten-2-one (Scheme 3B), and by electrochemical co-reformation of LA and VA (Scheme 3C).
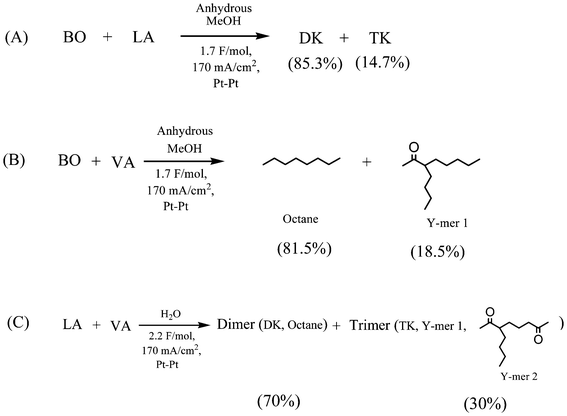 | ||
| Scheme 3 Electrolysis of 3-buten-2-one with LA (A), VA (B) in MeOH, and LA with VA (C) in H2O. | ||
The solvent composition is shown to affect the yield and ratio of DK and TK. A list of the product compositions and yields as a function of various H2O/MeOH ratios is given in Table 2. In general, it is observed that high water content has a negative impact on the current efficiency and the yield of DK. This is most likely due to a competitive electro-oxidation of hydroxide ions [HO−] and/or H2O molecules at the anode. For example, the yield of DK decreased from about 70% to roughly 35%, and the current efficiency decreased from about 84% to about 35%, as the water content increased to 77 mol%. The formation of TK showed the opposite trend, the ratio of TK/DK increased in proportion to the water content.
| [H2O]/[MeOH] (mol/mol) | Yield%a | TK/DKc | ||
|---|---|---|---|---|
| (DK) | (TK) | Total Yieldb | ||
| a Yield was determined by GC/FID using naphthalene as the internal standard. b Current efficiency (ec %) in brackets. c Yield ratio of product (TK) to (DK). | ||||
| MeOH | 68.3 | — | 68.3[83.3] | 0 |
| 20/80 | 68.2 | 5.1 | 73.3[83.6] | 0.07 |
| 35/65 | 57.3 | 8.6 | 65.9[48.1] | 0.15 |
| 50/50 | 42.5 | 11.0 | 53.5[39.3] | 0.26 |
| 70/30 | 41.9 | 14.4 | 56.3[35.3] | 0.39 |
| 77/23 | 34.9 | 20.3 | 55.2[34.2] | 0.58 |
| 84/16 | 32.7 | 21.6 | 54.2[35.3] | 0.64 |
| 90/10 | 32.8 | 20.6 | 53.3[35.3] | 0.66 |
| 95/5 | 33.1 | 22.1 | 55.2[33.4] | 0.68 |
| H2O | 24.4 | 18.4 | 42.8[31.9] | 0.75 |
2) Theoretical analysis of 5-acetyl-2,9-decanedione (TK) Li+ complex
The theoretical study of the solvation of lithium ions has been carried out in TK as a solvent. The optimized geometries, reaction parameters such as the standard enthalpy, entropy, and Gibbs energy changes at 298.15 K and 101325Pa, upon solvation Li+ in TK forms (Li+)n(TK)m, n = 0–2, m = 1–2, have been calculated by ab initio molecular orbital computation with the RHF/6-31G basis set. Configuration of (Li+)n(TK)m, n = 0–2, m = 1–2, lithium solvated complexes obtained by the geometrical optimization and the calculated electronic parameters such as atomic net charge of Li+ and the carbonyl O of the complexes (with fully geometrical optimization) are given in Fig. 4. Whereas the selected structure parameters (Li–O bond and CO bond distance) are presented in Table 3. The results of these computations indicate that Li+ is directly bonded to TK′s carbonyl oxygen atoms, which is consistent with results obtained with other carbonyl based common solvents, such as propylene carbonate (PC),2 ethylene carbonate (EC)3 and acetone23 (note, the PC and EC are common solvents for lithium ion batteries). Three possible complexes can form upon solvation of one lithium ion with one TK molecule, (Li+)1(TK)1: bidentates Li+ (TK)Li–O1O2, Li+ (TK)Li–O2O3 (Fig. 4b, 4c), and tridentate Li+ (TK) Li–O1O2O3 (Fig. 4d). The atomic net charges on the O atoms which coordinate Li+ become more negative from −0.547 to −0.734 for the bidentate (4b, 4c) and to −0.679 for the tridentate (4d) on average. It seems that Li+ coordination polarizes carbonyl CO bond, and thus increases the bond ionization characters and stretches the bond length of the CO bonds by 0.0185 Å on average, which is a similar phenomena of the binding characters of Li+ with acetone.23 The electronic oxygen–oxygen repulsion in the CO⋯Li⋯OC promotes a constrained bidentate or tridentate configuration. Thus, the average of Li–O bond distance of 1.814 Å for the bidentate, and 1.876 Å for the tridentate is slightly longer than that of line complexes Li+(acetone)2 (1.792 Å),3 Li+(PC)2 (1.774 Å).24
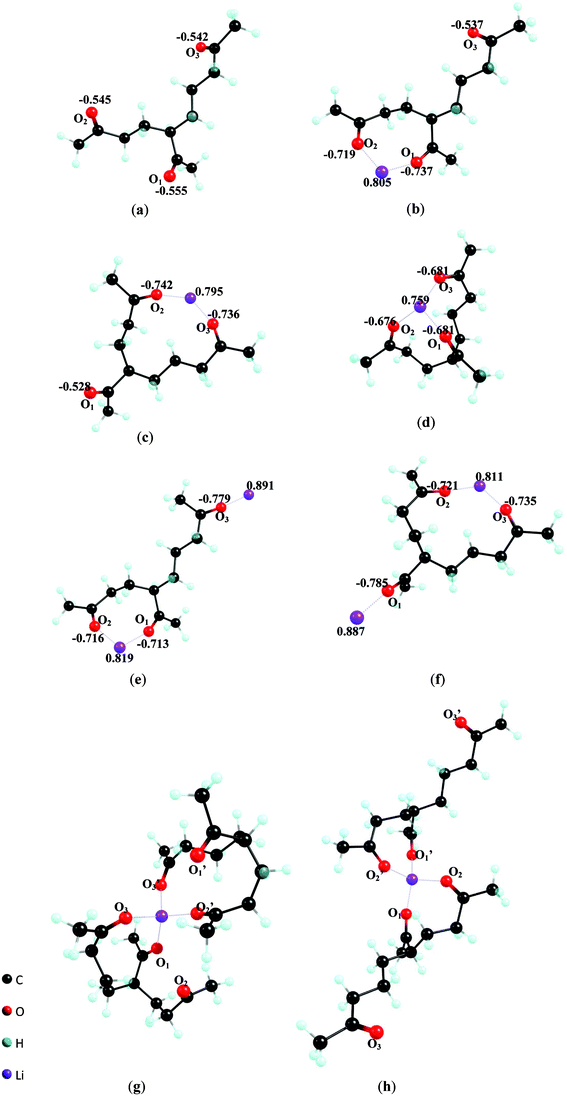 | ||
| Fig. 4 Optimized structure of (a) TK; (b) Li+(TK)Li–O1O2; (c) Li+(TK)Li–O2O3; (d) Li+(TK)Li–O1O2O3; (e) (Li+)2(TK) Li–O1O2, Li–O3; (f) (Li+)2(TK)Li–O2O3, Li–O1; (g) Li+(TK)Li–O1O3(TK)′Li–O2′O3′; (h) Li+(TK)Li–O1O2(TK)′Li–O1′O2′. | ||
O bond parametersa for (Li+)n(TK)mn = 0–2, m = 1–2
| Species | Distance of Li-carbonyl (Å) | Distance of CO (Å) |
||||
|---|---|---|---|---|---|---|
| Li–O1 | Li-O2 | Li–O3 | CO1 |
CO2 |
CO3 |
|
| a Calculated from ab initio molecular orbital computation using RHF/6-31G. | ||||||
| TK (4a) | - | - | - | 1.221 | 1.219 | 1.219 |
| Li+(TK)Li–O1O2 (4b) | 1.807 | 1.826 | - | 1.238 | 1.237 | 1.219 |
| Li+(TK)Li–O2O3(4c) | - | 1.807 | 1.814 | 1.218 | 1.241 | 1.238 |
| Li+((TK)Li–O1O2O3(4d) | 1.890 | 1.860 | 1.878 | 1.230 | 1.230 | 1.232 |
| (Li+)2(TK) Li–O1O2, Li–O3(4e) | 1.825 | 1.821 | 1.760 | 1.234 | 1.235 | 1.243 |
| (Li+)2(TK) Li–O2O3, Li–O1(4f) | 1.769 | 1.826 | 1.813 | 1.245 | 1.237 | 1.237 |
| Li+ (TK) Li–O1O3(TK)′Li–O2′O3′(4g) | 2.007 (-)′ | - (1.882)′ | 1.957 (1.983)′ | 1.230 (1.222)′ | 1.222 (1.233)′ | 1.229 (1.230)′ |
| Li+ (TK) Li–O1O2(TK)′Li–O1′O2′(4h) | 1.925 (1.925)′ | 1.958 (1.958)′ | - (-)′ | 1.229 (1.229)′ | 1.229 (1.229)′ | 1.219 (1.219)′ |
The atomic net charge on the Li+ is reduced from 1.0 to 0.805, 0.795 and 0.759 for complexes 4b–4d, respectively (Table 3). The transfer of electron density to the Li+ through the carbonyl O atoms of the TK molecule suggests a charge relaxation of Li+ upon the solvation and a higher degree of covalent bonding between Li+ and the carbonyl O.1 This is also evidenced by the heat of formation ΔH0298.15 K increasing in the order of 4b (−356.75 kJ mol−1) < 4c (−364.33 kJ mol−1) < 4d (− 404.84 kJ mol−1), Table 4. The Gibbs free energy of TK-Li complexes 4b–4d is −318.78 kJ mol−1, −320.23 kJ mol−1 and −354.52 kJ mol−1, respectively. Due to the collective effect of the three binding sites per TK molecule, the ΔG0298.15 K of complexes 4b–4d is ∼100 kJ mol−1 higher than complexes of lithium ion with one molecule of PC, EC, or actone (ΔG0298.15 K for Li+(PC)2, (Li+(EC),25 and Li+(acetone)3 is −188.28 kJ mol−1, −212.34kJ mol−1, −164.01 kJ mol−1, respectively). Based on these results, it can be assumed that the coordination of TK to the lithium ion would be favored over PC or EC (in a mixed solvent system as often used in the Li-ion battery).
| Reaction | ΔH°(298.15 K) (kJ mol−1) | ΔS°(298.15 K) (kJ mol−1) | ΔG°(298.15 K) (kJ mol−1) |
|---|---|---|---|
| Li+ + (TK) → Li+(TK)Li–O1O2 (4b) | −356.75 | −0.127 | −318.78 |
| Li+ + (TK) → Li+(TK)Li–O2O3 (4c) | −364.33 | −0.148 | −320.23 |
| Li+ + (TK) → Li+((TK)Li–O1O2O3(4d) | −404.84 | −0.169 | −354.52 |
| Li+ + 4b → (Li+)2(TK) Li–O1O2, Li–O3(4e) | −54.50 | −0.106 | −22.30 |
| Li+ + 4c → (Li+)2(TK) Li–O2O3, Li–O1(4f) | −32.01 | −0.104 | −0.98 |
| 4d+(TK)′ → Li+ (TK) Li–O1O3(TK)′Li–O2′O3′(4g) | −132.02 | −0.242 | −59.98 |
| 4b+(TK)′ → Li+ (TK) Li–O1O2(TK)′Li–O1′O2′(4h) | −178.27 | −0.157 | −131.51 |
| Li+ + (EC) → Li+(EC) | −243.24 | −0.097 | −214.20 |
| Li+ + (PC) → Li+(PC) | −249.95 | −0.100 | −220.20 |
Since TK possesses three carbonyl groups, it has the potential to bind more than one Li+. The free carbonyl O atoms on complexes 4b and 4c might solvate a second Li+ to form a (Li+)2(TK)1 complex such as (Li+)2(TK)Li–O1O2, Li–O3(4e), or (Li+)2(TK)Li–O2O3, Li–O1(4f). Upon binding a second Li+, the electron density on the complex is partially redistributed, which is demonstrated by the electron density loss of the Li+ and carbonyl O atoms in the O⋯Li⋯O structures (compared to the 4b or 4c structure), while the electron density gain of those of the second Li⋯O bond (e.g., the bond of Li⋯O3 in complex 4e (Li+)2(TK)Li–O1O2, Li–O3 compare to Li+). In these complexes, CO bonds in the bidentate structure are less polarized due to electron density loss of the lithium ion. Thus, the average distance of CO bonds contracts by ∼0.003 Å, however, the average distance of Li–O is slightly stretched by ∼0.007 Å for 4e and 0.009 Å for 4f.
In addition, the bond angle of Li⋯O3C in complex 4e is about 174.4°, in contrast to linear Li+(acetone) complexes (180°).23,26 Apparently, this due to electronic repulsion between the two Li+. The change in Gibbs free energy, ΔG, in reforming complex 4e from 4b is ∼−22.30 kJ mol−1, and reforming complex 4f from 4c is only about −0.98 kJ mol−1. The equilibrium constant of formation of 4e is ∼8.07 × 103, greater than the 4f (∼1.5), suggesting that the formation of complex 4e is a more favorable process than the formation complex 4f. It should be pointed out that solvating three lithium ions to form (Li+)3(TK)Li–O1, Li–O2, Li–O3 is thermodynamically unfavorable, since ΔG is greater than zero.
The solvation number of Li+ is also dependent on the steric features of the corresponding organic solvent. The favorable solvation number is reported to be four for PC or EC,2,3three for actone, two for diethylether23 and six for acetonitrile.27 In this study, we find that two TK molecules can be directly linked to a lithium ion using four carbonyl oxygen atoms, and another two tend to be liberated from the solvated complex. Thus, two possible formations are available, an asymmetrical tetrahedral complex of Li+ (TK) Li–O1O3(TK)′Li–O2′O3′ (Fig. 4g) in which Li+ is surrounded by carbonyl O1 and O3 from one TK, O2′ and O3′ from another TK′, and a symmetrical Li+ (TK) Li–O1O2(TK)′Li–O1′O2′ (Fig. 4h) where Li+ is surrounded by carbonyls O1and O2 from both TK molecules.
Conclusion
This work reports the discovery of a new compound, 5-acetyl-2,9-decanedione, that originates from electrochemical reformation of bio-derived levulinic acid. This new finding provides an additional (or new) understanding of the side reactions for the well-known Kolbe electrolysis reaction. The proposed mechanism could be a simple synthetic pathway which is an environmentally friendly “green sustainable” process that leads to the synthesis unique hierarchic “Y” structures with functional groups anchored at the end of each arm. This triketone unique “Y” structure of 5-acetyl-2,9-decanedione can dissolve lithium salts and be a potential solvent in lithium-ion battery. Theoretical computation results show the solvation energy (ΔH°) of lithium-TK complex (Li+)1(TK)1 reaches ∼350 kJ mol−1, greater than Li+ (PC) or Li+ (EC) by ∼100 kJ mol−1. The TK has also a capacity to solvate up to two Li+ per TK molecule. Further experimental and theoretical studies on this subject are under progress in this laboratory.Acknowledgements
Authors would like to thank Prof. Theodore Dibble and Mrs. Hongyi Hu for their help on the theoretical calculations and use of the GAUSSIAN 09 program package. The authors also wish to thank Prof. Stuart W. Tanenbaum and Prof. Neal M. Abrams for their helpful discussions and critical suggestions and Mr. D. Kiemle for his tremendous help with the GC-MS and NMR measurements.References
- R. J. Blint, J. Electrochem. Soc., 1995, 142, 696–702 CrossRef CAS.
- H. Ohtani, Y. Hirao, A. Ito, K. Tanaka and O. Hatozaki, J. Therm. Anal. Calorim, 2010, 99, 139–144 CrossRef CAS.
- S. Yanase and T. Oi, J. Nucl. Sci. Technol., 2002, 39, 1060–1064 CrossRef CAS.
- Y. Yamada, Y. Koyama, T. Abe and Z. Ogumi, J. Phys. Chem. C, 2009, 113, 8948–8953 CAS.
- R. T. Houk, E. V. Anslyn and J. F. Stanton, Org. Lett., 2006, 8, 3461–3464 CrossRef CAS PubMed.
- K. Amine, Q. Wang, D. R. Vissers, A. Zhang, N. A. A. Rossi and R. West, Electrochem. Comm., 2006, 8, 429–433 CrossRef CAS.
- M. B. Armand, Annu. Rev. Mater. Sci., 1986, 16, 245 CrossRef CAS.
- M. M. Bomgardner, Chemical & Engineering News, 2011, October 3, 22–23 Search PubMed.
- S. W. Fitzpatrick, The Biofine technology: a “bio-refinery” concept based on thermochemical conversion of cellulosic biomass, 2006, 921, 271–287 CAS.
- B. Girisuta, L. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- X. Liu and S. T. Yang, Process Biochem., 2006, 41, 801 CrossRef CAS.
- X. Liu, Y. Zhu and S. T. Yang, Enzyme. Microbiol. Technol., 2006, 38, 521 CrossRef CAS.
- T. Lund, D. Wayner, M. Jonsson, A. Larsen and K. Daasbjerg, J. Am. Chem. Soc, 2001, 123, 12590 CrossRef CAS PubMed.
- J. Yoshida, K. Kataoka, R. Horcajado and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed.
- H. Koble, Ann. Chem. Pharm., 1849, 69, 257 CrossRef.
- J. P. Coleman, R. Lines, J. Utley and B. C. Weedon, J.C.S. Perkin II, 1974, 1064 Search PubMed.
- V. A. V. Ginberg and B. Yu, Russian Journal of Electrochemistry, 1996, 32, 281 Search PubMed.
- H. J. Schafer, Top. Curr. Chem., 1990, 152, 91 CrossRef.
- R. A. Gaussian 09, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr.,J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian, Inc., Wallingford CT, 2009.
- E. Klocke, A. Matzeit, M. Gockeln and H. J. Schafer, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS.
- G. E. Svadkovskaya and S. A. Voitkevich, Russian Chemical Reviews, 1960, 29, 161 CrossRef.
- R. L. Jarek, T. D. Miles, M. L. Trester, S. C. Denson and S. K. Shin, J. Phys. Chem. A, 2000, 104, 2230–2237 CrossRef CAS.
- V. G. Gurjar, J. Appl. Electrochem., 1978, 8, 207–211 CrossRef CAS.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708–11718 CrossRef CAS PubMed.
- J. F. Hinton, A. Beeler, D. Harpool, R. W. Briggs and A. Pullman, Chem. Phys. Lett., 1977, 47, 411–415 CrossRef CAS.
- D. Spangberg and K. Hermansson, Chem. Phys., 2004, 300, 165–17 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |